Year 2007 Position Statement: Principles and Guidelines for Early Hearing Detection and Intervention Programs
Position statement
Joint Committee on Infant Hearing
Table of Contents
- The Position Statement
- JCIH 2007 Position Statement Updates
- I. Background
- II. Principles
- III. Guidelines for Early Hearing Detection and Intervention Programs
- A. Roles and Responsibilities
- B. Hearing Screening
- C. Confirmation of Hearing Loss in Infants Referred From UNHS
- D. Early Intervention
- E. Continued Surveillance, Screening, and Referral of Infants and Toddlers
- F. Protecting the Rights of Infants and Families
- G. Information Infrastructure
- H. Benchmarks and Quality Indicators
- IV. Current Challenges, Opportunities, and Future Directions
- V. Conclusions
- Acknowledgments
- Appendix 1. Risk Indicators Associated With Permanent Congenital, Delayed-Onset, or Progressive Hearing Loss in Childhood.
- Appendix 2. Algorithm for Hearing Screening. Available at: http://www.medicalhomeinfo.org/screening/Screen%20Materials/Algorithm.pdf
- References
- Notes
The Position Statement
The Joint Committee on Infant Hearing (JCIH) endorses early detection of and intervention for infants with hearing loss. The goal of early hearing detection and intervention (EHDI) is to maximize linguistic competence and literacy development for children who are deaf or hard of hearing. Without appropriate opportunities to learn language, these children will fall behind their hearing peers in communication, cognition, reading, and social-emotional development. Such delays may result in lower educational and employment levels in adulthood ( Holden & Diaz, 1998). To maximize the outcome for infants who are deaf or hard of hearing, the hearing of all infants should be screened no later than 1 month of age. Those not passing screening should have a comprehensive audiologic evaluation no later than 3 months of age. Infants with confirmed hearing loss should receive appropriate intervention no later than 6 months of age from health care and education professionals with expertise in hearing loss and deafness in infants and young children. Regardless of previous hearing-screening outcomes, all infants with or without risk factors should receive ongoing surveillance of communicative development beginning at 2 months of age during well-child visits in the medical home ( American Academy of Pediatrics [AAP] Medical Home Initiatives, 2002). EHDI systems should guarantee seamless transitions for infants and their families through this process.
JCIH 2007 Position Statement Updates
The following are highlights of updates made since the JCIH 2000 statement:
-
Definition of Targeted Hearing Loss
-
The definition has been expanded from congenital permanent bilateral, unilateral sensory, or permanent conductive hearing loss to include neural hearing loss (e.g., “auditory neuropathy/dyssynchrony”) in infants admitted to the neonatal intensive care unit (NICU).
-
-
Hearing Screening and Rescreening Protocols
-
Separate protocols are recommended for NICU and well-baby nurseries. NICU babies admitted for greater than 5 days are to have auditory brainstem response (ABR) included as part of their screening so that neural hearing loss will not be missed.
-
For infants who do not pass automated ABR in the NICU, referral should be made directly to an audiologist for rescreening and, when indicated, comprehensive evaluation including ABR.
-
For rescreening, a complete screening on both ears is recommended, even if only one ear failed the initial screening.
-
For readmissions in the first month of life for all infants (NICU or well baby) when there are conditions associated with potential hearing loss (e.g., hyperbilirubinemia requiring exchange transfusion or culture-positive sepsis), a repeat hearing screening is recommended before discharge.
-
-
Diagnostic Audiology Evaluation
-
Audiologists with skills and expertise in evaluating newborn and young infants with hearing loss should provide audiology diagnostic and auditory habilitation services (selection and fitting of amplification device).
-
At least one ABR test is recommended as part of a complete audiology diagnostic evaluation for children younger than 3 years for confirmation of permanent hearing loss.
-
The timing and number of hearing re-evaluations for children with risk factors should be customized and individualized depending on the relative likelihood of a subsequent delayed-onset hearing loss. Infants who pass the neonatal screening but have a risk factor should have at least one diagnostic audiology assessment by 24 to 30 months of age. Early and more frequent assessment may be indicated for children with cytomegalovirus (CMV) infection, syndromes associated with progressive hearing loss, neurodegenerative disorders, trauma, or culture-positive postnatal infections associated with sensorineural hearing loss; for children who have received ECMO or chemotherapy; and when there is caregiver concern or a family history of hearing loss.
-
For families who elect amplification, infants in whom permanent hearing loss is diagnosed should be fitted with an amplification device within 1 month of diagnosis.
-
-
Medical Evaluation
-
For infants with confirmed hearing loss, a genetics consultation should be offered to their families.
-
Every infant with confirmed hearing loss should be evaluated by an otolaryngologist with knowledge of pediatric hearing loss and have at least one examination to assess visual acuity by an ophthalmologist experienced in evaluating infants.
-
The risk factors for congenital and acquired hearing loss have been combined in a single list, rather than grouped by time of onset.
-
-
Early Intervention
-
All families of infants with any degree of bilateral or unilateral permanent hearing loss should be considered eligible for early intervention services.
-
There should be recognized central referral points of entry that ensure specialty services for infants with confirmed hearing loss.
-
Early intervention services for infants with confirmed hearing loss should be provided by professionals with expertise in hearing loss, including educators of the deaf, speech-language pathologists, and audiologists.
-
In response to a previous emphasis on “natural environments,” the committee recommends that both home-based and center-based intervention options should be offered.
-
-
Surveillance and Screening in the Medical Home
-
For all infants, regular surveillance of developmental milestones, auditory skills, parental concerns, and middle ear status should be performed in the medical home, consistent with the AAP pediatric periodicity schedule. All infants should have an objective standardized screening of global development with a validated assessment tool at 9, 18, and 24 to 30 months of age or at any time if the health care professional or family has concern.
-
Infants who do not pass the speech-language portion of a medical home global screening or for whom there is a concern regarding hearing or language should be referred for speech-language evaluation and audiology assessment.
-
-
Communication
-
The birth hospital, in collaboration with the state EHDI coordinator, should ensure that the hearing screening results are conveyed to the parents and the medical home.
-
Parents should be provided with appropriate follow-up and resource information, and hospitals should ensure that each infant is linked to a medical home.
-
Information at all stages of the EHDI process is to be communicated to the family in a culturally sensitive and understandable format.
-
Individual hearing screening information and audiology diagnostic and habilitation information should be promptly transmitted to the medical home and the state EHDI coordinator.
-
Families should be made aware of all communication options and available hearing technologies (presented in an unbiased manner). Informed family choice and desired outcome guide the decision-making process.
-
-
Information Infrastructure
-
States should implement data-management and tracking systems as part of an integrated child health information system to monitor the quality of EHDI services and provide recommendations for improving systems of care.
-
An effective link between health and education professionals is needed to ensure successful transition and to determine outcomes of children with hearing loss for planning and establishing public health policy.
-
I. Background
It has long been recognized that unidentified hearing loss at birth can adversely affect speech and language development as well as academic achievement and social-emotional development. Historically, moderate-to-severe hearing loss in young children was not detected until well beyond the newborn period, and it was not unusual for diagnosis of milder hearing loss and unilateral hearing loss to be delayed until school age.
In the late 1980s, Dr. C. Everett Koop, then U.S. Surgeon General, on learning of new technology, encouraged that detection of hearing loss be included in the Healthy People 2000 goals for the nation ( U.S. Department of Health and Human Services, 1991). In 1988, the Maternal and Child Health Bureau (MCHB), a division of the U.S. Health Resources and Services Administration (HRSA), funded pilot projects in Rhode Island, Utah, and Hawaii to test the feasibility of a universal statewide screening program to screen newborn infants for hearing loss before hospital discharge. The National Institutes of Health (NIH), through the National Institute on Deafness and Other Communication Disorders (NIDCD), issued in 1993 the “Consensus Statement on Early Identification of Hearing Impairment in Infants and Young Children” ( NIH, 1993). The statement concluded that all infants admitted to the NICU should be screened for hearing loss before hospital discharge and that universal screening should be implemented for all infants within the first 3 months of life ( U.S. Department of Health and Human Services, 1991). In its 1994 position statement, the JCIH endorsed the goal of universal detection of infants with hearing loss and encouraged continuing research and development to improve methods for identification of and intervention for hearing loss ( JCIH, 1994a, 1994b). The AAP released a statement recommending newborn hearing screening and intervention in 1999 ( AAP Task Force, 1999). In 2000, citing advances in screening technology, the JCIH endorsed the universal screening of all infants through an integrated, interdisciplinary system of EHDI ( JCIH, 2000a, 2000b). The Healthy People 2010 goals included an objective to “increase the proportion of newborns who are screened for hearing loss by one month, have audiologic evaluation by 3 months, and are enrolled in appropriate intervention services by 6 months” ( U.S. Department of Health and Human Services, 2000).
The ensuing years have seen remarkable expansion in newborn hearing screening. At the time of the NIH consensus statement, only 11 hospitals in the United States were screening more than 90% of their newborn infants. In 2000, through the support of Representative Jim Walsh (R-NY), Congress authorized HRSA to develop newborn hearing screening and follow-up services, the Centers for Disease Control and Prevention (CDC) to develop data and tracking systems, and NIDCD to support research in early hearing detection and intervention. By 2005, every state had implemented a newborn hearing-screening program, and approximately 95% of newborn infants in the United States were screened for hearing loss before hospital discharge. Congress recommended cooperation and collaboration among several federal agencies and advocacy organizations to facilitate and to support the development of state EHDI systems.
EHDI programs throughout the nation have demonstrated not only the feasibility of universal newborn hearing screening (UNHS) but also the benefits of early identification and intervention. There is a growing body of literature indicating that when identification and intervention occur no later than 6 months of age for newborn infants who are deaf or hard of hearing, the infants perform as much as 20 to 40 percentile points higher on school-related measures (vocabulary, articulation, intelligibility, social adjustment, and behavior; Yoshinaga-Itano, 1995, 2004; Yoshinaga-Itano, Coulter, & Thomson, 2000; Yoshinaga-Itano, Sedey, Coulter, & Mehl, 1998).
Still, many important challenges remain. Despite the fact that approximately 95% of newborn infants have their hearing screened in the United States, almost half of newborn infants who do not pass the initial screening fail to have appropriate follow-up to confirm the presence of a hearing loss and/or initiate appropriate early intervention services ( http://www.infanthearing.org/; http://www.cdc.gov/ncbddd/ehdi/; http://www.nidcd.nih.gov/health/).
State EHDI coordinators report system-wide problems, including failure to communicate information to families in a culturally sensitive and understandable format at all stages of the EHDI process, lack of integrated state data-management and tracking systems, and a shortage of facilities and personnel with the experience and expertise needed to provide follow-up for infants referred from newborn screening programs ( White, 2003). Available data indicate that a significant number of children who need further assessment do not receive appropriate follow-up evaluations. However, the outlook is improving as EHDI programs focus on the importance of strengthening follow-up and intervention.
II. Principles
All children with hearing loss should have access to resources necessary to reach their maximum potential. The following principles provide the foundation for effective EHDI systems and have been updated and expanded since the JCIH 2000 position statement.
-
All infants should have access to hearing screening using a physiologic measure no later than 1 month of age.
-
All infants who do not pass the initial hearing screening and the subsequent rescreening should have appropriate audiologic and medical evaluations to confirm the presence of hearing loss no later than 3 months of age.
-
All infants with confirmed permanent hearing loss should receive early intervention services as soon as possible after diagnosis but no later than 6 months of age. A simplified, single point of entry into an intervention system appropriate for children with hearing loss is optimal.
-
The EHDI system should be family centered with infant and family rights and privacy guaranteed through informed choice, shared decision making, and parental consent in accordance with state and federal guidelines. Families should have access to information about all intervention and treatment options and counseling regarding hearing loss.
-
The child and family should have immediate access to high-quality technology, including hearing aids, cochlear implants, and other assistive devices when appropriate.
-
All infants and children should be monitored for hearing loss in the medical home ( AAP Task Force, 2003). Continued assessment of communication development should be provided by appropriate professionals to all children with or without risk indicators for hearing loss.
-
Appropriate interdisciplinary intervention programs for infants with hearing loss and their families should be provided by professionals knowledgeable about childhood hearing loss. Intervention programs should recognize and build on strengths, informed choices, traditions, and cultural beliefs of the families.
-
Information systems should be designed and implemented to interface with electronic health records and should be used to measure outcomes and report the effectiveness of EHDI services at the patient, practice, community, state, and federal levels.
III. Guidelines for Early Hearing Detection and Intervention Programs
The 2007 guidelines were developed to update the 2000 JCIH position statement principles and to support the goals of universal access to hearing screening, evaluation, and intervention for newborn and young infants embodied in Healthy People 2010 ( U.S. Department of Health and Human Services, 2000). The guidelines provide current information on the development and implementation of successful EHDI systems.
Hearing screening should identify infants with specifically defined hearing loss on the basis of investigations of long-term, developmental consequences of hearing loss in infants, currently available physiologic screening techniques, and availability of effective intervention in concert with established principles of health screening ( AAP Committee, 2000; AAP Task Force, 2003; Fletcher, Fletcher, & Wagner, 1988; Sackett, Hayes, & Tugwell, 1991). Studies have demonstrated that current screening technologies are effective in identifying hearing loss of moderate and greater degree ( Norton et al., 2000). In addition, studies of children with permanent hearing loss indicate that moderate or greater degrees of hearing loss can have significant effects on language, speech, academic, and social-emotional development ( Carney & Moeller, 1998). High-risk target populations also include NICU infants, because research data indicate that this population is at highest risk of having neural hearing loss ( D'Agostino & Austin, 2004; Sininger, Hood, Starr, Berlin, & Picton, 1995; Starr, Sininger, & Pratt, 2000).
The JCIH, however, is committed to the goal of identifying all degrees and types of hearing loss in childhood and recognizes the developmental consequences of even mild degrees of permanent hearing loss. Recent evidence suggests, however, that current hearing screening technologies fail to identify some infants with mild forms of hearing loss ( Cone-Wesson et al., 2000; J. L. Johnson et al., 2005). Additionally, depending on the screening technology selected, infants with hearing loss related to neural conduction disorders or “auditory neuropathy/auditory dyssynchrony” may not be detected through a UNHS program. Although the JCIH recognizes that these disorders may result in delayed communication ( Berlin, Hood, Morlet, Rose, & Brashears, 2003; Doyle, Sininger, & Starr, 1998; Rance, 2005), currently recommended screening algorithms (i.e., use of otoacoustic emissions testing [OAE] alone) preclude universal screening for these disorders. Because these disorders typically occur in children who require NICU care ( D'Agostino & Austin, 2004), the JCIH recommends screening this group with the technology capable of detecting auditory neuropathy/dyssynchrony: automated ABR.
All infants, regardless of newborn hearing screening outcome, should receive ongoing monitoring for development of age-appropriate auditory behaviors and communication skills. Any infant who demonstrates delayed auditory and/or communication skills development, even if he or she passed newborn hearing screening, should receive audiologic evaluation to rule out hearing loss.
A. Roles and Responsibilities
The success of EHDI programs depends on families working in partnership with professionals as a well-coordinated team. The roles and responsibilities of each team member should be well defined and clearly understood. Essential team members are the birth hospital, families, pediatricians or primary health care professionals (i.e., the medical home), audiologists, otolaryngologists, speech-language pathologists, educators of children who are deaf or hard of hearing, and other early intervention professionals involved in delivering EHDI services ( American Speech-Language-Hearing Association, 1991, 1994). Additional services including genetics, ophthalmology, developmental pediatrics, service coordination, supportive family education, and counseling should be available ( Calderon, Bargones, & Sidman, 1998).
The birth hospital is a key member of the team. The birth hospital, in collaboration with the state EHDI coordinator, should ensure that parents and primary health care professionals receive and understand the hearing screening results, that parents are provided with appropriate follow-up and resource information, and that each infant is linked to a medical home ( AAP Medical Home Initiatives, 2002). The hospital ensures that hearing screening information is promptly transmitted to the medical home and appropriate data are submitted to the state EHDI coordinator.
The most important role for the family of an infant who is deaf or hard of hearing is to love, nurture, and communicate with the baby. From this foundation, families usually develop an urgent desire to understand and meet the special needs of their infant. Families gain knowledge, insight, and experience by accessing resources and through participation in scheduled early intervention appointments including audiologic, medical, habilitative, and educational sessions. This experience can be enhanced when families choose to become involved with parental support groups, individuals who are deaf or hard of hearing, and/or their children's deaf or hard-of-hearing peers. Informed family choices and desired outcomes guide all decisions for these children. A vital function of the family's role is ensuring direct access to communication in the home and the daily provision of language learning opportunities. Over time, the child benefits from the family's modeling of partnerships with professionals and advocating for their rights in all settings. The transfer of responsibilities from families to their child develops gradually and increases as their child matures, growing in independence and self-advocacy.
Pediatricians, family physicians, and other allied health care professionals, working in partnership with parents and other professionals such as audiologists, therapists, and educators, constitute the infant's medical home ( AAP Medical Home Initiatives, 2002). A medical home is defined as an approach to providing health care services where care is accessible, family centered, continuous, comprehensive, coordinated, compassionate, and culturally competent. The primary health care professional acts in partnership with parents in a medical home to identify and access appropriate audiology, intervention, and consultative services needed to develop a global plan of appropriate and necessary health and habilitative care for infants identified with hearing loss and infants with risk factors for hearing loss. All children undergo surveillance for auditory skills and language milestones. The infant's pediatrician, family physician, or other primary health care professional is in a position to advocate for the child and family ( AAP Committee, 2000; AAP Medical Home Initiatives, 2002).
An audiologist is a person who, by virtue of academic degree, clinical training, and license to practice, is qualified to provide services related to the prevention of hearing loss and the audiologic diagnosis, identification, assessment, and nonmedical and nonsurgical treatment of persons with impairment of auditory and vestibular function, and to the prevention of impairments associated with them. Audiologists serve in a number of roles. They provide newborn hearing screening program development, management, quality assessment, service coordination and referral for audiologic diagnosis, and audiologic treatment and management. For the follow-up component, audiologists provide comprehensive audiologic diagnostic assessment to confirm the existence of the hearing loss, ensure that parents understand the significance of the hearing loss, evaluate the infant for candidacy for amplification and other sensory devices and assistive technology, and ensure prompt referral to early intervention programs. For the treatment and management component, audiologists provide timely fitting and monitoring of amplification ( Pediatric Working Group, 1996). Other audiologists may provide diagnostic and auditory treatment and management services in the educational setting and provide a bridge between the child/family and the audiologist in the clinic setting as well as other service providers. Audiologists also provide services as teachers, consultants, researchers, and administrators.
Otolaryngologists are physicians whose specialty includes determining the etiology of hearing loss; identifying related risk indicators for hearing loss, including syndromes involving the head and neck; and evaluating and treating ear diseases. An otolaryngologist with knowledge of childhood hearing loss can determine whether medical and/or surgical intervention may be appropriate. When medical and/or surgical intervention is provided, the otolaryngologist is involved in the long-term monitoring and follow-up with the infant's medical home. The otolaryngologist provides information and participates in the assessment of candidacy for amplification, assistive devices, and surgical intervention, including reconstruction, bone-anchored hearing aids, and cochlear implantation.
Early intervention professionals are trained in a variety of academic disciplines, such as speech-language pathology, audiology, education of children who are deaf or hard of hearing, service coordination, or early childhood special education. All individuals who provide services to infants with hearing loss should have specialized training and expertise in the development of audition, speech, and language. Speech-language pathologists provide both evaluation and intervention services for language, speech, and cognitive-communication development. Educators of children who are deaf or hard of hearing integrate the development of communicative competence within a variety of social, linguistic, and cognitive/academic contexts. Audiologists may provide diagnostic and habilitative services within the individualized family service plan (IFSP) or school-based individualized education plan. To provide the highest quality of intervention, more than one provider may be required.
The care coordinator is an integral member of the EHDI team who facilitates the family's transition from screening to evaluation to early intervention ( AAP Committee, 2005). This individual must be a professional (e.g., social worker, teacher, nurse) who is knowledgeable about hearing loss. The care coordinator incorporates the family's preferences for outcomes into an IFSP as required by federal legislation. The care coordinator supports the family members in their choice of the infant's communicative development. Through the IFSP review, the infant's progress in language, motor, cognitive, and social-emotional development is monitored. The care coordinator assists the family in advocating for the infant's unique developmental needs.
The deaf and hard-of-hearing community includes members with direct experience with signed language, spoken language, hearing aid and cochlear implant use, and other communication strategies and technologies. Optimally, adults who are deaf or hard-of-hearing should play an integral part in the EHDI program. Both adults and children in the deaf and hard-of-hearing community can enrich the family's experience by serving as mentors and role models. Such mentors have experience in negotiating their way in a hearing world, raising infants or children who are deaf or hard of hearing, and providing families with a full range of information about communication options, assistive technology, and resources available in the community.
A successful EHDI program requires collaboration between a variety of public and private institutions and agencies assuming responsibility for specific components (e.g., screening, evaluation, intervention). Roles and responsibilities may differ from state to state. Each state has defined a lead coordinating agency with oversight responsibility. The lead coordinating agency in each state should be responsible for identifying the public and private funding sources available to develop, implement, and coordinate EHDI systems.
B. Hearing Screening
Multidisciplinary teams of professionals, including audiologists, physicians, and nursing personnel, are needed to establish the UNHS component of EHDI programs. All team members work together to ensure that screening programs are of high quality and are successful. An audiologist should be involved in each component of the hearing screening program, particularly at the level of statewide implementation and, whenever possible, at the individual hospital level. Hospitals and agencies should also designate a physician to oversee the medical aspects of the EHDI program.
Each team of professionals responsible for the hospital-based UNHS program should review the hospital infrastructure in relationship to the screening program. Hospital-based programs should consider screening technology (i.e., OAE or automated ABR testing); validity of the specific screening device; screening protocols, including the timing of screening relative to nursery discharge; availability of qualified screening personnel; suitability of the acoustical and electrical environments; follow-up referral criteria; referral pathways for follow-up; information management; and quality control and improvement. Reporting and communication protocols must be well defined and include the content of reports to physicians and parents, documentation of results in medical records, and methods for reporting to state registries and national data sets.
Physiologic measures must be used to screen newborns and infants for hearing loss. Such measures include OAE and automated ABR testing. Both OAE and automated ABR technologies provide noninvasive recordings of physiologic activity underlying normal auditory function, both are easily performed in neonates and infants, and both have been successfully used for UNHS ( Finitzo, Albright, & O'Neal, 1998; Mason & Hermann, 1998; Norton et al., 2000; Prieve et al., 2000; Vohr, Carty, Moore, & Letourneau, 1998). There are, however, important differences between the two measures. OAE measurements are obtained from the ear canal using a sensitive microphone within a probe assembly that records cochlear responses to acoustic stimuli. Thus, OAEs reflect the status of the peripheral auditory system extending to the cochlear outer hair cells. In contrast, ABR measurements are obtained from surface electrodes that record neural activity generated in the cochlea, auditory nerve, and brainstem in response to acoustic stimuli delivered via an earphone. Automated ABR measurements reflect the status of the peripheral auditory system, the eighth nerve, and the brainstem auditory pathway.
Both OAE and ABR screening technologies can be used to detect sensory (cochlear) hearing loss ( Norton et al., 2000); however, both technologies may be affected by outer or middle ear dysfunction. Consequently, transient conditions of the outer and middle ear may result in a “fail” screening test result in the presence of normal cochlear and/or neural function ( Doyle, Burggraaff, Fujikawa, Kim, & MacArthur, 1997). Moreover, because OAEs are generated within the cochlea, OAE technology cannot be used to detect neural (eighth nerve or auditory brainstem pathway) dysfunction. Thus, infants with neural conduction disorders or auditory neuropathy/dyssynchrony without concomitant sensory dysfunction will not be detected by OAE testing.
Some infants who pass newborn hearing screening will later demonstrate permanent hearing loss ( J. L. Johnson et al., 2005). Although this loss may reflect delayed-onset hearing loss, both ABR and OAE screening technologies will miss some hearing loss (e.g., mild or isolated frequency region losses).
Interpretive criteria for pass/fail outcomes should reflect clear scientific rationale and should be evidence-based ( M. D. Hyde, Sininger, & Don, 1998; M. L. Hyde, Davidson, & Alberti, 1991). Screening technologies that incorporate automated response detection are necessary to eliminate the need for individual test interpretation, to reduce the effects of screener bias or operator error on test outcome, and to ensure test consistency across infants, test conditions, and screening personnel ( Eilers, Miskiel, Ozdamar, Urbano, & Widen, 1991; Herrmann, Thornton, & Joseph, 1995; McFarland, Simmons, & Jones, 1980; Ozdamar, Delgado, Eilers, & Urbano, 1994; Pool & Finitzo, 1989). When statistical probability is used to make pass/fail decisions, as is the case for OAE and automated ABR screening devices, the likelihood of obtaining a pass outcome by chance alone is increased when screening is performed repeatedly ( Benjamini & Yekutieli, 2005; Hochberg & Benjamini, 1990; Zhang, Chung, & Oldenburg, 1999). This principle must be incorporated into the policies of rescreening.
There are no national standards for the calibration of OAE or ABR instrumentation. Compounding this, there is a lack of uniform performance standards. Manufacturers of hearing-screening devices do not always provide sufficient supporting evidence to validate the specific pass/fail criteria and/or automated algorithms utilized in their instruments ( Gravel et al., 2005). In the absence of national standards, audiologists must obtain normative data for the instruments and protocols they use.
The JCIH recognizes that there are important issues differentiating screening performed in the well-baby nursery from that performed in the NICU. Although the goals in each nursery are the same, numerous methodologic and technological issues must be considered in program design and pass/fail criteria.
Screening Protocols in the Well-Baby Nursery
Many inpatient well-baby screening protocols provide one hearing screening and, when necessary, a repeat screening no later than the time of discharge from the hospital, using the same technology both times. Use of either technology in the well-baby nursery will detect peripheral (conductive and sensory) hearing loss of 40 dB or greater ( Norton et al., 2000). When automated ABR is used as the single screening technology, neural auditory disorders can also be detected ( Sininger, Abdala, & Cone-Wesson, 1997). Some programs use a combination of screening technologies (OAE testing for the initial screening, followed by automated ABR for rescreening; i.e., 2-step protocol; NIH, 1993), to decrease the fail rate at discharge and the subsequent need for outpatient follow-up ( Arehart, Yoshinaga-Itano, Thomson, Gabbard, & Brown, 1998; Finitzo et al., 1998; Gravel et al., 2000; Mason & Hermann, 1998; Mehl & Thomson, 1998; Vohr et al., 1998). Using this approach, infants who do not pass an OAE screening but subsequently pass an automated ABR are considered a screening “pass.” Infants in the well-baby nursery who fail automated ABR should not be rescreened by OAE and “passed,” because such infants are presumed to be at risk of having a subsequent diagnosis of auditory neuropathy/dyssynchrony.
Screening Protocols in the NICU
A NICU is defined as a facility in which a neonatologist provides primary care for the infant. Newborn units are divided into categories as follows:
-
Level I: basic care, well-baby nurseries
-
Level II: specialty care by a neonatologist for infants at moderate risk of serious complications
-
Level III: a unit that provides both specialty and subspecialty care including the provision of life support (mechanical ventilation)
A total of 120 level-II NICUs and 760 level-III NICUs have been identified in the United States by survey, and infants who have spent time in the NICU represent 10% to 15% of the newborn population ( Stark & AAP, 2004).
The JCIH 2007 position statement includes neonates at risk of having neural hearing loss (auditory neuropathy/auditory dyssynchrony) in the target population to be identified in the NICU ( Berg, Spitzer, Towers, Bartosiewicz, & Diamond, 2005; Shapiro, 2003; Starr, Picton, Sininger, Hood, & Berlin, 1996), because there is evidence that neural hearing loss results in adverse communication outcomes ( Sininger, Abdala, & Cone-Wesson, 1997; Sininger et al., 1995). Consequently, the JCIH recommends ABR technology as the only appropriate screening technique for use in the NICU. For infants who do not pass automated ABR testing in the NICU, referral should be made directly to an audiologist for rescreening and, when indicated, comprehensive evaluation, including diagnostic ABR, rather than for general outpatient rescreening.
Conveying Test Results
Screening results should be conveyed immediately to families so they understand the outcome and the importance of follow-up when indicated. To facilitate this process for families, primary health care professionals should work with EHDI team members to ensure the following:
-
Communications with parents are confidential and presented in a caring and sensitive manner, preferably face-to-face.
-
Educational materials are developed and disseminated to families that provide accurate information at an appropriate reading level and in a language they are able to comprehend.
-
Parents are informed in a culturally sensitive and understandable manner that their infant did not pass screening and informed about the importance of prompt follow-up. Before discharge, parents should be offered an appointment for follow-up testing.
To facilitate this process for primary care physicians, EHDI systems should ensure the following:
-
Medical professionals receive the results of the screening test (pass, did not pass, or missed) as documented in the hospital medical record.
-
Medical professionals receive communication directly from the hospital screening program regarding each infant in their care who did not pass or is missed and recommendations for follow-up.
Outpatient Rescreening for Infants Who Do Not Pass the Birth Admission Screening
Many well-baby screening protocols will choose to incorporate an outpatient rescreening within 1 month of hospital discharge to minimize the number of infants referred for follow-up audiologic and medical evaluation. The outpatient rescreening should include the testing of both ears, even if only one ear failed the inpatient screening.
Outpatient screening no later than 1 month of age should also be available to infants who were discharged before receiving the birth admission screening or who were born outside a hospital or birthing center. State EHDI coordinators should be aware of some of the following situations under which infants may be lost to the UNHS system:
-
Home births and other out-of-hospital births: States should develop a mechanism to systematically offer newborn hearing screening for all out-of-hospital births.
-
Across state border births: States should develop written collaborative agreements among neighboring states for sharing hearing screening results and follow-up information.
-
Hospital missed screenings: When infants are discharged before the hearing screening is performed, a mechanism should be in place for the hospital to contact the family and arrange for an outpatient hearing screening.
-
Transfers to in-state or out-of-state hospitals: Discharge and transfer forms should contain the information of whether a hearing screening was performed and the results of any screening. The recipient hospital should complete a hearing screening if not previously performed or if there is a change in medical status or a prolonged hospitalization.
-
Readmits: For readmissions in the first month of life when there are conditions associated with potential hearing loss (e.g., hyperbilirubinemia requiring exchange transfusion or culture-positive sepsis), a screening ABR should be performed before discharge.
Additional mechanisms for states to share hearing screening results and other medical information include (a) incorporating the hearing screening results in a statewide child health information system and (b) providing combined metabolic screening and hearing screening results to the primary care physician.
C. Confirmation of Hearing Loss in Infants Referred From UNHS
Infants who meet the defined criteria for referral should receive follow-up audiologic and medical evaluations with fitting of amplification devices, as appropriate, no later than 3 months of age. Once hearing loss is confirmed, coordination of services should be expedited by the infant's medical home and Part C coordinating agencies for early intervention services, as authorized by the Individuals with Disabilities Education Act, following the EHDI algorithm developed by the AAP ( Appendix 1; AAP Task Force, 2003).
Audiologic Evaluation
Comprehensive audiologic evaluation of newborn and young infants who fail newborn hearing screening should be performed by audiologists experienced in pediatric hearing assessment. The initial audiologic test battery to confirm a hearing loss in infants must include physiologic measures and, when developmentally appropriate, behavioral methods. Confirmation of an infant's hearing status requires a test battery of audiologic test procedures to assess the integrity of the auditory system in each ear, to estimate hearing sensitivity across the speech frequency range, to determine the type of hearing loss, to establish a baseline for further monitoring, and to provide information needed to initiate amplification device fitting. Comprehensive assessment should be performed on both ears even if only one ear failed the screening test.
Evaluation: Birth to 6 Months of Age
For infants from birth to a developmental age of approximately 6 months, the test battery should include a child and family history, an evaluation of risk factors for congenital hearing loss, and a parental report of the infant's responses to sound. The audiologic assessment should include the following:
-
Child and family history
-
A frequency-specific assessment of the ABR using air-conducted tone bursts and bone-conducted tone bursts when indicated. When permanent hearing loss is detected, frequency-specific ABR is needed to determine the degree and configuration of hearing loss in each ear for fitting of amplification devices.
-
Click-evoked ABR using both condensation and rarefaction single-polarity stimulus, if there are risk indicators for neural hearing loss (auditory neuropathy/auditory dyssynchrony) such as hyperbilirubinemia or anoxia, to determine whether a cochlear microphonic is present ( Rance, 2005). Furthermore, because some infants with neural hearing loss have no risk indicators, any infant who demonstrates "no response" on ABR elicited by tone burst stimuli must be evaluated by a click-evoked ABR, as previously described.
-
Distortion product or transient evoked otoacoustic emissions.
-
Tympanometry using a 1000-Hz probe tone.
-
Clinician observation of the infant's auditory behavior as a cross-check, in conjunction with electrophysiologic measures. Behavioral observation alone is not adequate for determining whether hearing loss is present in this age group, nor is it adequate for the fitting of amplification devices.
Evaluation: 6 to 36 Months of Age
For subsequent testing of infants and toddlers at developmental ages of 6 to 36 months, the confirmatory audiologic test battery includes the following:
-
Child and family history
-
Parental report of auditory and visual behaviors and communication milestones.
-
Behavioral audiometry (either visual reinforcement or conditioned-play audiometry, depending on the child's developmental level), including pure-tone audiometry across the frequency range for each ear and speech detection and recognition measures.
-
OAE testing.
-
Acoustic immittance measures (tympanometry and acoustic reflex thresholds).
-
ABR, if responses to behavioral audiometry are not reliable or if ABR has not been performed in the past.
Other Audiologic Test Procedures
At this time, there is insufficient evidence for use of the auditory steady state response as the sole measure of auditory status in newborn and infant populations ( Stapells, Gravel, & Martin, 1995). Auditory steady state response is a new evoked potential test that can accurately measure auditory sensitivity beyond the limits of other test methods. It can determine frequency specific thresholds from 250 Hz to 8 kHz. Clinical research is investigating its potential use in the standard pediatric diagnostic test battery.
Similarly, there are insufficient data for routine use of acoustic middle ear muscle reflexes in the initial diagnostic assessment of infants younger than 4 months ( Keefe, Gorga, Neely, Zhao, & Vohr, 2003). Both tests could be used to supplement the battery or could be included at older ages. Emerging technologies, such as broad-band reflectance, may be used to supplement conventional measures of middle ear status (tympanometry and acoustic reflexes) as the technology becomes more widely available ( Keefe et al., 2003).
Medical Evaluation
Every infant with confirmed hearing loss and/or middle ear dysfunction should be referred for otologic and other medical evaluation. The purpose of these evaluations is to determine the etiology of hearing loss, to identify related physical conditions, and to provide recommendations for medical/surgical treatment as well as referral for other services. Essential components of the medical evaluation include clinical history, family history of childhood-onset permanent hearing loss, identification of syndromes associated with early- or late-onset permanent hearing loss, a physical examination, and indicated radiologic and laboratory studies (including genetic testing). Portions of the medical evaluation, such as urine culture for CMV, a leading cause of hearing loss, might even begin in the birth hospital, particularly for infants spending time in the NICU ( Boppana et al., 2005; Nagy, Endreffy, Streitman, Pinter, & Pusztai, 2004; Roizen, 1999).
Pediatrician/Primary Care Physician
The infant's pediatrician or other primary health care professional is responsible for monitoring the general health, development, and well-being of the infant. In addition, the primary care physician must assume responsibility to ensure that the audiologic assessment is conducted on infants who do not pass screening and must initiate referrals for medical specialty evaluations necessary to determine the etiology of the hearing loss. Middle-ear status should be monitored, because the presence of middle-ear effusion can further compromise hearing. The primary care physician must partner with other specialists, including the otolaryngologist, to facilitate coordinated care for the infant and family. Because 30% to 40% of children with confirmed hearing loss will demonstrate developmental delays or other disabilities, the primary care physician should closely monitor developmental milestones and initiate referrals related to suspected disabilities ( Karchmer & Allen, 1999). The medical home algorithm for management of infants with either suspected or proven permanent hearing loss is provided in Appendix 2 ( AAP Task Force, 2003).
The pediatrician or primary care physician should review every infant's medical and family history for the presence of risk indicators that require monitoring for delayed-onset or progressive hearing loss and should ensure that an audiologic evaluation is completed for children at risk of hearing loss at least once by 24 to 30 months of age, regardless of their newborn screening results ( J. L. Johnson et al., 2005). Infants with specific risk factors, such as those who received ECMO therapy and those with CMV infection, are at increased risk of delayed-onset or progressive hearing loss ( Fligor, Neault, Mullen, Feldman, & Jones, 2005; Fowler et al., 1992; Madden et al., 2005; Rivera et al., 2002) and should be monitored closely. In addition, the primary care physician is responsible for ongoing surveillance of parent concerns about language and hearing, auditory skills, and developmental milestones of all infants and children regardless of risk status, as outlined in the pediatric periodicity schedule published by the AAP Committee on Practice and Ambulatory Medicine ( 2000).
Children with cochlear implants may be at increased risk of acquiring bacterial meningitis compared with children in the general U.S. population ( Reefhuis et al., 2003). The CDC recommends that all children with, and all potential recipients of, cochlear implants follow specific recommendations for pneumococcal immunization that apply to cochlear implant users and that they receive age-appropriate Haemophilus influenzae type b vaccines.
Recommendations for the timing and type of pneumococcal vaccine vary with age and immunization history and should be discussed with a health care professional ( CDC, 2003).
Otolaryngologist
Otolaryngologists are physicians and surgeons who diagnose, treat, and manage a wide range of diseases of the head and neck, specializing in treating hearing and vestibular disorders. They perform a full medical diagnostic evaluation of the head and neck, ears, and related structures, including a comprehensive history and physical examination, leading to a medical diagnosis and appropriate medical and surgical management. Often, a hearing or balance disorder is an indicator of, or related to, a medically treatable condition or an underlying systemic disease. Otolaryngologists work closely with other dedicated professionals, including physicians, audiologists, speech-language pathologists, educators, and others, in caring for patients with hearing, balance, voice, speech, developmental, and related disorders.
The otolaryngologist's evaluation includes a comprehensive history to identify the presence of risk factors for early-onset childhood permanent hearing loss, such as family history of hearing loss, having been admitted to the NICU for >5 days, and having received ECMO (see Appendix 1; Morzaria, Westerberg, & Kozak, 2005; Preciado et al., 2005).
A complete head and neck examination for craniofacial anomalies should document defects of the auricles, patency of the external ear canals, and status of the eardrum and middle ear structures. Atypical findings on eye examination, including irises of two different colors or abnormal positioning of the eyes, may signal a syndrome that includes hearing loss. Congenital permanent conductive hearing loss may be associated with craniofacial anomalies seen in disorders such as Crouzon disease, Klippel-Feil syndrome, and Goldenhar syndrome ( Nance, 2003). The assessment of infants with these congenital anomalies should be coordinated with a clinical geneticist.
In large population studies, at least 50% of congenital hearing loss has been designated as hereditary, and nearly 600 syndromes and 125 genes associated with hearing loss have already been identified ( Brookhouser, Worthington, & Kelly, 1994; Nance, 2003). The evaluation, therefore, should include a review of family history of specific genetic disorders or syndromes, including genetic testing for gene mutations such as GJB2 (connexin-26), and syndromes commonly associated with early-onset childhood sensorineural hearing loss ( Appendix 1; Denoyelle et al., 1999; Nance, 2003; Nance & Kearsey, 2004; Santos et al., 2005). As the widespread use of newly developed conjugate vaccines decreases the prevalence of infectious etiologies such as measles, mumps, rubella, H. influenzae type b, and childhood meningitis, the percentage of each successive cohort of early-onset hearing loss attributable to genetic etiologies can be expected to increase, prompting recommendations for early genetic evaluations. Approximately 30% to 40% of children with hearing loss have associated disabilities, which can be of importance in patient management. The decision to obtain genetic testing is dependent on informed family choice in conjunction with standard confidentiality guidelines ( NIDCD, 1999).
In the absence of a genetic or established medical cause, a computed tomography scan of the temporal bones may be performed to identify cochlear abnormalities, such as Mondini deformity or an enlarged vestibular aqueduct, which have been associated with progressive hearing loss. Temporal bone imaging studies may also be used to assess potential candidacy for surgical intervention, including reconstruction, bone-anchored hearing aid, and cochlear implantation. Recent data have shown that some children with electrophysiologic evidence suggesting auditory neuropathy/dyssynchrony may have an absent or abnormal cochlear nerve that may be detected on magnetic resonance imaging ( Buchman et al., 2006).
Historically, an extensive battery of laboratory and radiographic studies was routinely recommended for newborn infants and children with newly diagnosed sensorineural hearing loss. However, emerging technologies for the diagnosis of genetic and infectious disorders have simplified the search for a definitive diagnosis, obviating the need for costly diagnostic evaluations in some instances ( Morzaria et al., 2005; Preciado et al., 2004, 2005).
If, after an initial evaluation, the etiology remains uncertain, an expanded multidisciplinary evaluation protocol including electrocardiography, urinalysis, testing for CMV, and further radiographic studies is indicated. The etiology of neonatal hearing loss, however, may remain uncertain in as many as 30% to 40% of children. Once hearing loss is confirmed, medical clearance for hearing aids and initiation of early intervention should not be delayed while this diagnostic evaluation is in process. Careful longitudinal monitoring to detect and promptly treat coexisting middle ear effusions is an essential component of ongoing otologic management of these children.
Other Medical Specialists
The medical geneticist is responsible for the interpretation of family history data, the clinical evaluation and diagnosis of inherited disorders, the performance and assessment of genetic tests, and the provision of genetic counseling. Geneticists or genetic counselors are qualified to interpret the significance and limitations of new tests and to convey the current status of knowledge during genetic counseling. All families of children with confirmed hearing loss should be offered and may benefit from a genetics evaluation and counseling. This evaluation can provide families with information on etiology of hearing loss, prognosis for progression, associated disorders (e.g., renal, vision, cardiac), and likelihood of recurrence in future offspring. This information may influence parents' decision making regarding intervention options for their child.
Every infant with a confirmed hearing loss should have an evaluation by an ophthalmologist to document visual acuity and rule out concomitant or late-onset vision disorders, such as Usher syndrome ( Holden-Pitt & Diaz, 1998; D. H. Johnson, 1999). Indicated referrals to other medical subspecialists, including developmental pediatricians, neurologists, cardiologists, and nephrologists, should be facilitated and coordinated by the primary health care professional.
D. Early Intervention
Before newborn hearing screening was instituted universally, children with severe to profound hearing loss, on average, completed the 12th grade with a 3rd- to 4th-grade reading level and language levels of a 9- to 10-year-old hearing child ( Traxler, 2000). In contrast, infants and children with mild to profound hearing loss who are identified in the first 6 months of life and provided with immediate and appropriate intervention have significantly better outcomes than later-identified infants and children in vocabulary development ( Mayne, Yoshinaga-Itano, & Sedey, 1998; Mayne, Yoshinaga-Itano, Sedey, & Carey, 1998), receptive and expressive language ( Pipp-Siegel, Sedey, VanLeeuwen, & Yoshinaga-Itano, 2003; Yoshinaga-Itano et al., 1998), syntax ( Yoshinaga-Itano, Coulter, & Thomson, 2001), speech production ( Apuzzo & Yoshinaga-Itano, 1995; Yoshinaga-Itano & Apuzzo, 1998a, 1998b; Yoshinaga-Itano et al., 2000), and social-emotional development ( Yoshinaga-Itano, 2001). Children enrolled in early intervention within the first year of life have also been shown to have language development within the normal range of development at 5 years of age ( Calderon et al., 1998; Moeller, 2000).
Therefore, according to federal guidelines, once any degree of hearing loss is diagnosed in a child, a referral should be initiated to an early intervention program within 2 days of confirmation of hearing loss (CFA 303.321d). The initiation of early intervention services should begin as soon as possible after diagnosis of hearing loss, but no later than 6 months of age. Even when the hearing status is not determined to be the primary disability, the family and child should have access to intervention with a provider knowledgeable about hearing loss ( Kennedy, McCann, Campbell, Kimm, & Thornton, 2005).
UNHS programs have been instituted throughout the United States for the purpose of preventing the significant and negative effects of hearing loss on the cognitive, language, speech, auditory, social-emotional, and academic development of infants and children. To achieve this goal, hearing loss must be identified as quickly as possible after birth, and appropriate early intervention must be available to all families and infants with permanent hearing loss. Some programs have demonstrated that most children with hearing loss and no additional disabilities can achieve and maintain language development within the typical range of children with normal hearing ( Moeller, 2000; Yoshinaga-Itano, Coulter, & Thomson, 2000, 2001; Yoshinaga-Itano et al., 1998). Because these studies are descriptive and not causal studies, the efficacy of specific components of intervention cannot be separated from the total provision of comprehensive services. Thus, the family-centered philosophy, the intensity of services, the experience and training of the provider, the method of communication, the curricula, the counseling procedures, the parent support and advocacy, and the deaf and hard-of-hearing support and advocacy are all variables with unknown effects on the overall outcomes of any individual child. The key component of providing quality services is the expertise of the provider specific to hearing loss. These services may be provided in the home or in a center, or a combination of the two locations.
The term “intervention services” is used to describe any type of habilitative, rehabilitative, or educational program provided to individuals with hearing loss. In some cases with mild hearing losses, amplification technology may be the only service provided. Some parents choose only developmental assessment or occasional consultation, such as parents with infants who have unilateral hearing losses. Children with high-frequency losses and normal hearing in the low frequencies may only be seen by a speech-language pathologist, and those with significant bilateral sensorineural hearing losses might be seen by an educator of the deaf and receive additional services.
Principles of Early Intervention
To ensure informed decision making, parents of infants with newly diagnosed hearing loss should be offered opportunities to interact with other families who have infants or children with hearing loss as well as adults and children who are deaf or hard of hearing. In addition, parents should also be offered access to professional, educational, and consumer organizations and provided with general information on child development, language development, and hearing loss. A number of principles and guidelines have been developed that offer a framework for quality early intervention service delivery systems for children who are deaf or hard of hearing and their families ( Bodner-Johnson & Sass-Lehrer, 2003). Foundational characteristics of developing and implementing early intervention programs include a family-centered approach, culturally responsive practices, collaborative professional-family relationships and strong family involvement, developmentally appropriate practice, interdisciplinary assessment, and community-based provision of services.
Designated Point of Entry
States should develop a single point of entry into intervention specific for hearing impairment to ensure that, regardless of geographic location, all families who have infants or children with hearing loss receive information about a full range of options regarding amplification and technology, communication and intervention, and accessing appropriate counseling services. This state system, if separate from the state's Part C system, should integrate and partner with the state's Part C program. Parental consent must be obtained, according to state and federal requirements, to share the IFSP information with providers and transmit data to the state EHDI coordinator.
Regular Developmental Assessment
To ensure accountability, individual, community, and state health and educational programs should assume the responsibility for coordinated, ongoing measurement and improvement of EHDI process outcomes. Early-intervention programs must assess the language, cognitive skills, auditory skills, speech, vocabulary, and social-emotional development of all children with hearing loss at 6-month intervals during the first 3 years of life, using assessment tools standardized on children with normal hearing and norm-referenced assessment tools that are appropriate to measure progress in verbal and visual language.
The primary purpose of regular developmental monitoring is to provide valuable information to parents about the rate of their child's development as well as programmatic feedback concerning curriculum decisions. Families also become knowledgeable about expectations and milestones of typical development of hearing children. Studies have shown that valid and reliable documentation of developmental progress is possible through parent questionnaires, analysis of videotaped conversational interactions, and clinically administered assessments ( Arehart et al., 1998; Moeller, 2000; Yoshinaga-Itano, 1995, 2001, 2003a, 2003b, 2004; Yoshinaga-Itano & Abdala de Uzcategui, 2001; Yoshinaga-Itano & Apuzzo, 1998a, 1998b; Yoshinaga-Itano, Coulter, & Thomson, 2000, 2001; Yoshinaga-Itano & Sedey, 1998; Yoshinaga-Itano et al., 1998). Documentation of developmental progress should be provided on a regular basis to parents and, with parental release of information, to the medical home and audiologist. Although criterion-referenced checklists may provide valuable information for establishing intervention strategies and goals, these assessment tools alone are not sufficient for parents and intervention professionals to determine whether a child's developmental progress is comparable with his or her hearing peers.
Opportunities for Interaction With Other Parents of Children With Hearing Loss
Intervention professionals should seek to involve parents at every level of the EHDI process and develop true and meaningful partnerships with parents. To reflect the value of the contributions that selected parents make to development and program components, these parents should be paid as contributing staff members. Parent representatives should be included in all advisory board activities. In many states, parents have been integral and often have taken leadership roles in the development of policy, resource material, communication mechanisms, mentoring and advocacy opportunities, dissemination of information, and interaction with the deaf community and other individuals who are deaf or hard of hearing. Parents, often in partnership with individuals who are deaf and hard of hearing, have also participated in the training of professionals. They should be participants in the regular assessment of program services to ensure ongoing improvement and quality assurance.
Opportunities for Interaction With Individuals Who Are Deaf or Hard of Hearing
Intervention programs should include opportunities for involvement of individuals who are deaf or hard of hearing in all aspects of EHDI programs. Because intervention programs serve children with mild to profound, unilateral or bilateral, permanent conductive, and sensory or neural hearing disorders, role models who are deaf or hard of hearing can be significant assets to an intervention program. These individuals can serve on state EHDI advisory boards and can be trained as mentors for families and children with hearing loss who choose to seek their support. Almost all families choose at some time during their early childhood programs to seek out both adults and child peers with hearing loss. Programs should ensure that these opportunities are available and can be delivered to families through a variety of communications means, such as Web sites, e-mail, newsletters, videos, retreats, picnics and other social events, and educational forums for parents.
Provision of Communication Options
Research studies thus far of early-identified infants with hearing loss have not found significant differences in the developmental outcomes by method of communication when measured at 3 years of age ( Moeller, 2000; Yoshinaga-Itano, 1995, 2004; Yoshinaga-Itano & Abdala de Uzcategui, 2001; Yoshinaga-Itano & Apuzzo, 1998a, 1998b; Yoshinaga-Itano, Coulter, & Thomson, 2000, 2001; Yoshinaga-Itano & Sedey, 1998; Yoshinaga-Itano et al., 1998). Therefore, a range of options should be offered to families in a nonbiased manner. In addition, there are reports of children with successful outcomes for each of the different methods of communication. The choice is a dynamic process on a continuum, differs according to the individual needs of each family, and can be adjusted as necessary on the basis of a child's rate of progress in developing communication skills. Programs need to provide families with access to skilled and experienced early intervention professionals to facilitate communication and language development in the communication option chosen by the family.
Skills of the Early Intervention Professional
All studies with successful outcomes reported for early-identified children who are deaf or hard of hearing have intervention provided by specialists trained in parent-infant intervention services ( Calderon, 2000; Moeller, 2000; Yoshinaga-Itano et al., 1998). Early intervention programs should develop mechanisms to ensure that early intervention professionals have special skills necessary for providing families with the highest quality of service specific to children with hearing loss. Professionals with a background in deaf education, audiology, and speech-language pathology will typically have the skills needed for providing intervention services. Professionals should be highly qualified in their respective fields and should be skilled communicators who are knowledgeable and sensitive to the importance of enhancing families' strengths and supporting their priorities. When early intervention professionals have knowledge of the principles of adult learning, it increases their success with parents and other professionals.
Quality of Intervention Services
Children with confirmed hearing loss and their families have the right to prompt access to quality intervention services. For newborn infants with confirmed hearing loss, enrollment into intervention services should begin as soon after hearing loss confirmation as possible, and no later than 6 months of age. Successful early intervention programs (a) are family centered, (b) provide families with unbiased information on all options regarding approaches to communication, (c) monitor development at 6-month intervals using norm-referenced instruments, (d) include individuals who are deaf or hard of hearing, (e) provide services in a natural environment in the home or in the center, (f) offer high-quality service regardless of where the family lives, (g) obtain informed consent, (h) are sensitive to cultural and language differences and provide accommodations as needed, and (i) conduct annual surveys of parent satisfaction.
Intervention for Special Populations of Infants and Young Children
Developmental monitoring should also occur at regular 6-month intervals for special populations of children with hearing loss, including minimal and mild bilateral hearing loss ( Bess, Dodd-Murphy, & Parker, 1998), unilateral hearing loss ( Bess & Tharpe, 1984, 1986), and neural hearing loss ( Sininger et al., 1995), because these children are at risk of having speech and language delay. Research findings indicate that approximately one third of children with permanent unilateral loss experience significant language and academic delays ( Bess & Tharpe, 1984, 1986).
Audiologic Habilitation
Most infants and children with bilateral hearing loss and many with unilateral hearing loss benefit from some form of personal amplification ( Pediatric Working Group, 1996). If the family chooses personal amplification for their infant, hearing aid selection and fitting should occur within 1 month of initial confirmation of hearing loss even when additional audiologic assessment is ongoing. Audiologic habilitation services should be provided by an audiologist experienced with these procedures. Delay between confirmation of the hearing loss and fitting of an amplification device should be minimized ( American Speech-Language-Hearing Association, 2004; Arehart et al., 1998).
Hearing aid fitting proceeds optimally when the results of physiologic audiologic assessment including diagnostic ABR, OAE and tympanometry and medical examination are in accord. For infants below a developmental age of six months, hearing aid selection will be based on physiologic measures alone. Behavioral threshold assessment using visual reinforcement audiometry should be obtained as soon as possible to cross-check and augment physiologic findings ( American Academy of Audiology, 2003).
The goal of amplification device fitting is to provide the infant with maximum access to all of the acoustic features of speech within an intensity range that is safe and comfortable. That is, amplified speech should be comfortably above the infant's sensory threshold, but below the level of discomfort across the speech frequency range for both ears. To accomplish this in infants, amplification device selection, fitting, and verification should be based on a prescriptive procedure that incorporates individual real-ear measures that account for each infant's ear canal acoustics and hearing loss ( Pediatric Working Group, 1996). Validation of the benefits of amplification, particularly for speech perception, should be examined in the clinical setting as well as in the child's typical listening environments. Complementary or alternative technology, such as FM systems or cochlear implants, may be recommended as the primary and/or secondary listening device, depending on the degree of the infant's hearing loss, the goals of auditory habilitation, the infant's acoustic environments, and the family's informed choices ( JCIH, 2000a, 2000b). Monitoring of amplification, as well as the long-term validation of the appropriateness of the individual habilitation program, requires ongoing audiologic assessment along with electroacoustic, real-ear, and functional checks of the hearing instruments. As the hearing loss becomes more specifically defined through audiologic assessments and as the child's ear canal acoustics change with growth, refinement of the individual prescriptive hearing aid gain and output targets is necessary. Monitoring also includes periodic validation of communication, social-emotional, and cognitive development and, later, academic performance to ensure that progress is commensurate with the child's abilities. It is possible that infants and young children with measurable residual “hearing” (auditory responses) and well-fit amplification devices may fail to develop auditory skills necessary for successful oral communication. Ongoing validation of the amplification device is accomplished through interdisciplinary evaluation and collaboration with the early intervention team and family.
Cochlear implantation should be given careful consideration for any child who appears to receive limited benefit from a trial with appropriately fitted hearing aids. According to U.S. Food and Drug Administration guidelines, infants with profound bilateral hearing loss are candidates for cochlear implantation at 12 months of age and children with bilateral severe hearing loss are eligible at 24 months of age. The presence of developmental conditions (e.g., developmental delay, autism) in addition to hearing loss should not, as a rule, preclude the consideration of cochlear implantation for an infant or child who is deaf. Benefits from hearing aids and cochlear implants in children with neural hearing loss have also been documented. The benefit of acoustic amplification for children with neural hearing loss is variable ( Rance, 2005; Rance, Cone-Wesson, Wunderlich, & Dowell, 2002). Thus, a trial fitting is indicated for infants with neural hearing loss until the usefulness of the fitting can be determined. Neural hearing loss is a heterogeneous condition; the decision to continue or discontinue use of hearing aids should be made on the basis of the benefit derived from amplification. Use of cochlear implants in neural hearing loss is growing, and positive outcomes have been reported for many children ( Rance, 2005).
Infants and young children with unilateral hearing loss should also be assessed for appropriateness of hearing aid fitting. Depending on the degree of residual hearing in unilateral loss, a hearing aid may or may not be indicated. Use of “contralateral routing of signals” amplification for unilateral hearing loss in children is not recommended ( American Academy of Audiology, 2003). Research is currently underway to determine how to best manage unilateral hearing loss in infants and young children.
The effect of otitis media with effusion (OME) is greater for infants with sensorineural hearing loss than for those with normal cochlear function ( Brookhouser, Worthington, & Kelly, 1994). Sensory or permanent conductive hearing loss is compounded by additional transient conductive hearing loss associated with OME. OME further reduces access to auditory cues necessary for the development of spoken English. OME also negatively affects the prescriptive targets of the hearing aid fitting, decreasing auditory awareness and requiring adjustment of the amplification characteristics. Prompt referral to either the primary care physician or an otolaryngologist for treatment of persistent OME is indicated in infants with sensorineural hearing loss ( Rosenfeld et al., 2004). Definitive resolution of OME should never delay the fitting of an amplification device ( Brookhouser et al., 1994; Diefendorf & Gravel, 1996).
Medical and Surgical Intervention
Medical intervention is the process by which a physician provides medical diagnosis and direction for medical and/or surgical treatment options for hearing loss and/or related medical disorder(s) associated with hearing loss. Treatment varies from the removal of cerumen and the treatment of OME to long-term plans for reconstructive surgery and assessment of candidacy for cochlear implants. If necessary, surgical treatment of malformation of the outer and middle ears, including bone-anchored hearing aids, should be considered in the intervention plan for infants with permanent conductive or mixed hearing loss when a child reaches an appropriate age.
Communication Assessment and Intervention
Language is acquired with greater ease during certain sensitive periods of infant and toddler development ( Clark, 1994; Mahshie, 1995; Sharma et al., 2004). The process of language acquisition includes learning the precursors of language, such as the rules pertaining to selective attention and turn taking ( Carney & Moeller, 1998; Kuhl et al., 1997; Kuhl, Williams, Lacerda, Stevens, & Lindblom, 1992). Cognitive, social, and emotional development are influenced by the acquisition of language. Development in these areas is synergistic. A complete language evaluation should be performed at regular intervals for infants and toddlers with hearing loss. The evaluation should include an assessment of oral, manual, and/or visual mechanisms as well as cognitive abilities.
A primary focus of language intervention is to support families in fostering the communication abilities of their infants and toddlers who are deaf or hard of hearing ( Carney & Moeller, 1998). Spoken and/or sign language development should be commensurate with the child's age and cognitive abilities and should include acquisition of phonologic (for spoken language), visual/spatial/motor (for signed language), morphologic, semantic, syntactic, and pragmatic skills, depending on the family's preferred mode of communication.
Early intervention professionals should follow family-centered principles to assist in developing communicative competence of infants and toddlers who are deaf or hard of hearing ( Baker-Hawkins & Easterbooks, 1994; Bamford, 1998; Fischer, 1994). Families should be provided with information specific to language development and access to peer and language models as well as family-involved activities that facilitate language development of children with normal hearing and children who are hard of hearing or deaf ( Marschark, 1997; Thompson, 1994). Depending on family choices, families should be offered access to children and adults with hearing loss who are appropriate and competent language models. Information on spoken language and signed language, such as American Sign Language ( Pollack, Goldberg, & Caleffe-Schenck, 1997) and cued speech, should be provided.
E. Continued Surveillance, Screening, and Referral of Infants and Toddlers
Appendix 1 presents 11 risk indicators associated with either congenital or delayed-onset hearing loss. A single list of risk indicators is presented in the current JCIH statement because there is significant overlap among those indicators associated with congenital/neonatal hearing loss and those associated with delayed-onset/acquired or progressive hearing loss. Heightened surveillance of all infants with risk indicators is, therefore, recommended. There is a significant change in the definition of risk indicator 3, which has been modified from NICU stay >48 hours to NICU stay >5 days. Consistent with JCIH 2000, the 2007 position statement recommends use of risk indicators for hearing loss for three purposes. Historically, the first use of risk indicators is for the identification of infants who should receive audiologic evaluation but who live in geographic locations (e.g., developing nations, remote areas) where universal hearing screening is not yet available ( Barbi et al., 2006; Barrenas, Jonsson, Tuvemo, Hellstrom, & Lundgren, 2005; Cone-Wesson et al., 2000; D'Agostino & Austin, 2004; Davis & Hind, 2003; Fligor et al., 2005; Jacobson & Jacobson, 2004; J. L. Johnson et al., 2005; JCIH, 2000a, 2000b; Mestan, Marks, Hecox, Huo, & Schreiber, 2005; Norton et al., 2000; Robertson, Tyebkhan, Peliowski, Etches, & Cheung, 2006; Vohr et al., 2000). This use has become less common as a result of the expansion of UNHS. The second purpose of risk indicator identification is to help identify infants who pass the neonatal screening but are at risk of developing delayed-onset hearing loss and, therefore, should receive ongoing medical, speech and language, and audiologic surveillance. Third, the risk indicators are used to detect infants who may have passed neonatal screening but have mild forms of permanent hearing loss ( J. L. Johnson et al., 2005).
Because some important indicators, such as family history of hearing loss, may not be determined during the course of UNHS ( Nance, 2003; White, 2003), the presence of all risk indicators for acquired hearing loss should be determined in the medical home during early well-baby visits. Risk indicators that are marked with an asterisk in Appendix 1 are of greater concern for delayed-onset hearing loss. Early and more frequent assessment may be indicated for children with CMV infection ( Barbi et al., 2006; Nance, Lim, & Dodson, 2006; Pass, Fowler, Boppana, Britt, & Stagno, 2006), syndromes associated with progressive hearing loss ( Nance, 2003), neurodegenerative disorders ( Nance, 2003), trauma ( Lew et al., 2004; Vartialnen, Karjalainen, & Karja, 1985; Zimmerman, Ganzel, Windmill, Nazar, & Phillips, 1993), or culture-positive postnatal infections associated with sensorineural hearing loss ( Arditi et al., 1998; Roizen, 2003); for children who have received ECMO ( Fligor et al., 2005) or chemotherapy ( Bertolini et al., 2004); and when there is caregiver concern or a family history of hearing loss ( AAP Committee, 2000).
For all infants with and without risk indicators for hearing loss, developmental milestones, hearing skills, and parent concerns about hearing, speech, and language skills should be monitored during routine medical care consistent with the AAP periodicity schedule.
The JCIH has determined that the previously recommended approach to follow-up of infants with risk indicators for hearing loss only addressed children with identifiable risk indicators and failed to consider the possibility of delayed-onset hearing loss in children without identifiable risk indicators. In addition, concerns were raised about feasibility and cost associated with the JCIH 2000 recommendation for audiologic monitoring of all infants with risk indicators at 6-month intervals. Because approximately 400,000 infants are cared for annually in NICUs in the United States, and the JCIH 2000 recommendation included audiology assessments at 6-month intervals from 6 months to 36 months of age for all infants admitted to a NICU for >48 hours, an unreasonable burden was placed on both providers of audiology services and families. In addition, there was no provision for identification of delayed-onset hearing loss in infants without an identifiable risk indicator. Data from 2005 for 12,388 infants discharged from NICUs in the National Perinatal Information Network indicate that 52% of infants were discharged within the first 5 days of life and these infants were significantly less likely to have an identified risk indicator for hearing loss other than NICU stay. Therefore, the JCIH 2007 recommends an alternative, more inclusive strategy of surveillance of all children within the medical home based on the pediatric periodicity schedule. This protocol will permit the detection of children with either missed neonatal or delayed-onset hearing loss, irrespective of the presence or absence of a high-risk indicator.
The JCIH recognizes that an optimal surveillance and screening program within the medical home would include the following:
-
At each visit, consistent with the AAP periodicity schedule, infants should be monitored for auditory skills, middle ear status, and developmental milestones (surveillance). Concerns elicited during surveillance should be followed by administration of a validated global screening tool ( AAP Council on Children, 2006). A validated global screening tool is administered at 9, 18, and 24 to 30 months to all infants or, if there is physician or parental concern about hearing or language, sooner ( AAP Council on Children, 2006).
-
If an infant does not pass the speech-language portion of the global screening in the medical home or if there is physician or caregiver concern about hearing or spoken language development, the child should be referred immediately for further evaluation by an audiologist and a speech-language pathologist for a speech and language evaluation with validated tools ( AAP Council on Children, 2006).
-
Once hearing loss is diagnosed in an infant, siblings who are at increased risk of having hearing loss should be referred for audiologic evaluation ( Fortnum & Davis, 1997; Nance & Kearsey, 2004; Orzan et al., 1999; White, 2003).
-
All infants with a risk indicator for hearing loss ( Appendix 1), regardless of surveillance findings, should be referred for an audiologic assessment at least once by 24 to 30 months of age. Children with risk indicators highly associated with delayed-onset hearing loss, such as having received ECMO or having CMV infection, should have more frequent audiologic assessments.
-
All infants for whom the family has significant concerns regarding hearing or communication should be promptly referred for an audiologic and speech-language assessment.
-
A careful assessment of middle ear status (using pneumatic otoscopy and/or tympanometry) should be completed at all well-child visits, and children with persistent middle ear effusion lasting 3 months or longer should be referred for otologic evaluation ( AAP Subcommittee, 2004).
F. Protecting the Rights of Infants and Families
Each agency or institution involved in the EHDI process shares responsibility for protecting infant and family rights in all aspects of UNHS, including access to information about potential benefits and risks in the family's native language, input into decision making, and confidentiality ( NIDCD, 1999). Families should receive information about childhood hearing loss in easily understood language. Families have the right to accept or decline hearing screening or any follow-up care for their newborn infant within the statutory regulations, just as they have for any other screening or evaluation procedures or intervention.
EHDI data merit the same level of confidentiality and security afforded all other health care and education information in practice and law. The infant's family has the right to confidentiality of the screening and follow-up assessments and the acceptance or rejection of suggested intervention(s). In compliance with federal and state laws, mechanisms should be established that ensure parental release and approval of all communications regarding the infant's test results, including those to the infant's medical home and early intervention coordinating agency and programs. The Health Insurance Portability and Accountability Act (Pub. L. No. 104-191 [1996]) regulations permit the sharing of health information among health care professionals.
G. Information Infrastructure
In the 2000 position statement, the JCIH recommended development of uniform state registries and national information databases incorporating standardized methodology, reporting, and system evaluation ( JCIH, 2000a, 2000b). EHDI information systems are to provide for the ongoing and systematic collection, analysis, and interpretation of data in the process of measuring and reporting associated program services (e.g., screening, evaluation, diagnosis, and/or intervention). These systems are used to guide activities, planning, implementation, and evaluation of programs and to formulate research hypotheses.
EHDI information systems are generally authorized by legislators and implemented by public health officials. These systems vary from a simple system collecting data from a single source to electronic systems that receive data from many sources in multiple formats. The number and variety of systems will likely increase with advances in electronic data interchange and integration of data, which will also heighten the importance of patient privacy, data confidentiality, and system security. Appropriate agencies and/or officials should be consulted for any projects regarding public health surveillance ( CDC, 2003).
Federal and state agencies are collaborating in the standardization of data definitions to ensure the value of data sets and to prevent misleading or unreliable information. Information management is used to improve services to infants and their families; to assess the quantity and timeliness of screening, evaluation, and enrollment into intervention; and to facilitate collection of demographic data on neonatal and infant hearing loss.
The JCIH endorses the concept of a limited national database to permit documentation of the demographics of neonatal hearing loss, including prevalence and etiology across the United States. The information obtained from the information management system should assist both the primary health care professional and the state health agency in measuring quality indicators associated with program services (e.g., screening, diagnosis, and intervention). The information system should provide the measurement tools to determine the degree to which each process is stable and sustainable and conforms to program benchmarks. Timely and accurate monitoring of relevant quality measures is essential.
Since 1999, the CDC and the Directors of Speech and Hearing Programs in State Health and Welfare Agencies (DSHPSHWA) have collected annual aggregate EHDI program data needed to address the national EHDI goals. In 1999, a total of 22 states provided data for the DSHPSHWA survey. Participation had increased to 48 states, 1 territory, and the District of Columbia in 2003. However, many programs have been unable to respond to all the questions on the survey because of lack of a statewide comprehensive data-management and reporting system.
The Government Performance and Results Act (GPRA) of 1993 (Pub. L. No. 103-62) requires that federal programs establish measurable goals approved by the U.S. Office of Management and Budget (OMB) that can be reported as part of the budgetary process, thus linking future funding decisions with performance. The HRSA has modified its reporting requirements for all grant programs. The following GPRA measures must be reported to the OMB by the MCHB annually for the EHDI program:
-
the number of infants screened for hearing loss before discharge from the hospital;
-
the number of infants with a confirmed with hearing loss no later than 3 months of age;
-
the number of infants enrolled in a program of early intervention no later than 6 months of age;
-
the number of infants with confirmed or suspected hearing loss referred to an ongoing source of comprehensive health care (i.e., medical home);
-
the number of children with nonsyndromic hearing loss who have developmentally appropriate language and communication skills at school entry.
The following is one GPRA measure that must be reported to the OMB by the CDC annually for the EHDI program:
-
the percentage of newborn infants with a positive screening result for hearing loss who are subsequently lost to follow-up.
EHDI programs have made tremendous gains in their ability to collect, analyze, and interpret data in the process of measuring and reporting associated program services. However, only a limited number of EHDI programs are currently able to accurately report the number of infants screened, evaluated, and enrolled in intervention, the age of time-related objectives (e.g., screening by 1 month of age), and the severity or laterality of hearing loss. This is complicated by the lack of data standards and by privacy issues within the regulations of the Family Educational Rights and Privacy Act of 1974 (Pub. L. No. 93-380).
Given the current lack of standardized and readily accessible sources of data, the CDC EHDI program, in collaboration with the DSHPSHWA, developed a revised survey to obtain annual EHDI data from states and territories in a consistent manner to assess progress toward meeting the national EHDI goals and the Healthy People 2010 Objectives. In October 2006, the OMB, which is responsible for reviewing all government surveys, approved the new Early Hearing Detection and Intervention Hearing Screening and Follow-Up Survey. To facilitate this effort, the CDC EHDI Data Committee is establishing the minimum data elements and definitions needed for information systems to be used to assess progress toward the national EHDI goals.
The JCIH encourages the CDC and HRSA to continue their efforts to identify barriers and explore possible solutions with EHDI programs to ensure that children in each state who seek hearing-related services in states other than where they reside receive all recommended screening and follow-up services. EHDI systems should also be designed to promote the sharing of data regarding early hearing loss through integration and/or linkage with other child health information systems. The CDC currently provides funds to integrate the EHDI system with other state/territorial screening, tracking, and surveillance programs that identify children with special health care needs. Grantees of the MCHB are encouraged to link hearing screening data with such child health data sets as electronic birth certificate, vital statistics, birth defects registries, metabolic or newborn dried “blood spot” screening, immunization registries, and others.
To promote the best use of public health resources, EHDI information systems should be periodically evaluated, and such evaluations should include recommendations for improving quality, efficiency, and usefulness. The appropriate evaluation of public health surveillance systems becomes paramount as these systems adapt to revise case definitions, address new health-related events, adopt new information technology, ensure data confidentiality, and assess system security ( CDC, 2003).
Currently, federal sources of systems support include Title V block grants to states for maternal and child health care services, Title XIX (Medicaid) federal and state funds for eligible children, and competitive U.S. Department of Education personnel preparation and research grants. The NIDCD provides grants for research related to early identification and intervention for children who are deaf or hard of hearing ( Roush et al., 2004).
Universities should assume responsibility for special-track, interdisciplinary, professional education programs for early intervention for infants and children with hearing loss. Universities should also provide training in family systems, the grieving process, cultural diversity, auditory skill development, and deaf culture. There is a critical need for in-service and preservice training of professionals related to EHDI programs, which is particularly acute for audiologists and early interventionists with expertise in hearing loss. This training will require increased and sustained funding for personnel preparation.
H. Benchmarks and Quality Indicators
The JCIH supports the concept of regular measurements of performance and recommends routine monitoring of these measures for interprogram comparison and continuous quality improvement. Performance benchmarks represent a consensus of expert opinion in the field of newborn hearing screening and intervention. The benchmarks are the minimal requirements that should be attained by high-quality EHDI programs. Frequent measures of quality permit prompt recognition and correction of any unstable component of the EHDI process ( Agency for Health Care Policy and Research, 1995).
Quality Indicators for Screening
-
Percentage of all newborn infants who complete screening by 1 month of age. Recommended benchmark is >95% (age correction for preterm infants is acceptable).
-
Percentage of all newborn infants who fail initial screening and fail any subsequent rescreening before comprehensive audiologic evaluation. Recommended benchmark is <4%.
Quality Indicators for Confirmation of Hearing Loss
-
Of infants who fail initial screening and any subsequent rescreening, the percentage who complete a comprehensive audiologic evaluation by 3 months of age. Recommended benchmark is 90%.
-
For families who elect amplification, the percentage of infants with confirmed bilateral hearing loss receiving amplification devices within 1 month of confirmation of hearing loss. Recommended benchmark is 95%.
Quality Indicators for Early Intervention
-
For infants with confirmed hearing loss who qualify for Part C, the percentage for whom parents have signed an IFSP no later than 6 months of age. Recommended benchmark is 90%.
-
For children with acquired or late-identified hearing loss, the percentage for whom parents have signed an IFSP within 45 days of the diagnosis. Recommended benchmark is 95%.
-
The percentage of infants with confirmed hearing loss who receive the first developmental assessment using standardized assessment protocols (not criterion reference checklists) for language, speech, and nonverbal cognitive development no later than 12 months of age. Recommended benchmark is 90%.
IV. Current Challenges, Opportunities, and Future Directions
Challenges
All of the following listed challenges are considered important for the future development of successful EHDI systems:
-
Too many children are lost between the failed screening and the rescreening and between the failed rescreening and the diagnostic evaluation.
-
There is a shortage of professionals with skills and expertise in both pediatrics and hearing loss, including audiologists, deaf educators, speech-language pathologists, early intervention professionals, and physicians.
-
There is often a lack of timely referral for diagnosis of, and intervention for, suspected hearing loss in children.
-
Consistent and stable state and federal funding is needed for program sustainability.
-
When compared with services provided for adults, pediatric services in all specialties are poorly reimbursed.
-
Access to uniform Part C services is inadequate among states and within states.
-
There is a lack of integrated state data-management and tracking systems.
-
Demographics and cultural diversity are rapidly changing.
-
Funding for hearing aids, loaner programs, cochlear implants, and FM systems is needed.
-
There is a lack of specialized services for children with multiple disabilities and hearing loss.
-
Children may not qualify for services (state Part C guidelines) before demonstrating language delays (prevention model vs. deficit model).
-
Children may not qualify for assistive technology (prevention model vs. deficit model).
-
There is a lack of in-service education for key professionals.
-
There are regulatory barriers to sharing information among providers and among states.
-
No national standards exist for the calibration of OAE or ABR instrumentation, and there is a lack of uniform performance standards.
Opportunities for System Development and Research
-
Establish programs to ensure the development of communication for infants and children with all degrees and types of hearing loss, allowing them access to all educational, social, and vocational opportunities throughout their life span.
-
Develop improved, rapid, reliable screening technology designed to differentiate specific types of hearing loss.
-
Develop and validate screening technologies for identifying minimal hearing loss.
-
Develop state data-management systems with the capacity for the accurate determination of the prevalence for delayed-onset or progressive hearing loss.
-
Develop state data-tracking systems to follow infants with suspected and confirmed hearing loss through individual state EHDI programs.
-
Track the certification credentials of the service providers for children with confirmed hearing loss receiving Part C early intervention services and early childhood special education.
-
Track genetic, environmental, and pharmacologic factors that contribute to hearing loss, thus allowing for tailored prevention and intervention strategies.
-
Continue to refine electrophysiologic diagnostic techniques, algorithms, and equipment to enable frequency-specific threshold assessment for use with very young infants.
-
Continue to refine techniques to improve the selection and fitting of appropriate amplification devices in infants and young children.
-
Conduct translational research pertaining to young children with hearing loss, in particular, genetic, diagnostic, and outcomes studies.
-
Initiate prospective population-based studies to determine the prevalence and natural history of auditory neural conduction disorders.
-
Conduct efficacy studies to determine appropriate early intervention strategies for infants and children with all degrees and types of hearing loss.
-
Conduct additional studies on the efficacy of intervention for infants and children who receive cochlear implants younger than 2 years.
-
Conduct additional studies on the efficacy of hearing aid use in infants and children younger than 2 years.
-
Conduct additional studies of the auditory development of children who have appropriate amplification devices in early life.
-
Expand programs within health, social service, and education agencies associated with early intervention and Head Start programs to accommodate the needs of the increasing numbers of early-identified children.
-
Adapt education systems to capitalize on the abilities of children with hearing loss who have benefited from early identification and intervention.
-
Develop genetic and medical procedures that will determine more rapidly the etiology of hearing loss.
-
Ensure transition from Part C (early intervention) to Part B (education) in ways that encourage family participation and ensure minimal disruption of child and family services.
-
Study the effects of parents' participation in all aspects of early intervention.
-
Test the utility of a limited national dataset and develop nationally accepted indicators of EHDI system performance.
-
Encourage the identification and development of centers of expertise where specialized care is provided in collaboration with local service providers.
-
Obtain the perspectives of individuals who are deaf or hard of hearing in developing policies regarding medical and genetic testing and counseling for families who carry genes associated with hearing loss ( Brick, 1999).
V. Conclusions
Since the JCIH 2000 statement, tremendous and rapid progress has been made in the development of EHDI systems as a major public health initiative. The percentage of infants screened annually in the United States has increased from 38% to 95%. The collaboration at all levels of professional organizations, federal and state government, hospitals, medical homes, and families has contributed to this remarkable success. New research initiatives to develop more sophisticated screening and diagnostic technology, improved digital hearing aid and FM technologies, speech-processing strategies in cochlear implants, and early intervention strategies continue. Major technological breakthroughs have been made in facilitating the definitive diagnosis of both genetic and nongenetic etiologies of hearing loss. In addition, outcomes studies to assess the long-term outcomes of special populations, including infants and children with mild and unilateral hearing loss, neural hearing loss, and severe or profound hearing loss managed with cochlear implants, are providing information on the individual and societal impact and the factors that contribute to an optimized outcome. It is apparent, however, that there are still serious challenges to be overcome and system barriers to be conquered to achieve optimal EHDI systems in all states in the next 5 years. Follow-up rates remain poor in many states, and funding for amplification in children is inadequate. Funding to support outcome studies is necessary to guide intervention and to determine factors other than hearing loss that affect child development. The ultimate goal, to optimize communication, social, academic, and vocational outcomes for each child with permanent hearing loss, must remain paramount.
Acknowledgments
The Year 2007 Position Statement was developed by the Joint Committee on Infant Hearing (JCIH). Joint committee member organizations and their respective representatives who prepared this statement include (in alphabetical order): the Alexander Graham Bell Association for the Deaf and Hard of Hearing (Jackie Busa, BA; and Judy Harrison, MA); the American Academy of Audiology (Jodi Chappell; Christine Yoshinaga-Itano, PhD; Alison Grimes, AuD; and Yvonne Siniger, PhD); the American Academy of Otolaryngology-Head and Neck Surgery (Patrick E. Brookhouser, MD; and Stephen Epstein, MD); the American Academy of Pediatrics (Albert Mehl, MD; and Betty Vohr, MD [Chairperson, March 2005-present]); the American Speech-Language-Hearing Association (Judith Gravel, PhD [Chair, March 2003-March 2005]; Jackson Roush, PhD; and Judith Widen, PhD); the Council on Education of the Deaf, whose member organizations include the Alexander Graham Bell Association for the Deaf and Hard of Hearing, American Society for Deaf Children, Conference of Educational Administrators of Schools and Programs for the Deaf, Convention of American Instructors of the Deaf, National Association of the Deaf, and Association of College Educators of the Deaf and Hard of Hearing (Beth S. Benedict, PhD; and Bobbie Scoggins, EdD); and the Directors of Speech and Hearing Programs in State Health and Welfare Agencies (Michelle King, MS, AuD; Linda Pippins, MCD; and David H. Savage, MSC). Ex officio contributors to the JCIH include Jill Ackermann, MS; Amy Gibson, MS, RN; and Thomas Tonniges, MD (American Academy of Pediatrics); and Pamela Mason, MEd (American Speech-Language-Hearing Association). We also acknowledge the contribution of John Eichwald, MA; and Irene Forsman, MS, RN.
Joint committee member organizations that adopt this statement include (in alphabetical order): the Alexander Graham Bell Association for the Deaf and Hard of Hearing, the American Academy of Audiology, the American Academy of Otolaryngology-Head and Neck Surgery, the American Academy of Pediatrics, the American Speech-Language-Hearing Association, the Council on Education of the Deaf (see individual organizations listed above), and the Directors of Speech and Hearing Programs in State Health and Welfare Agencies.
Appendix 1. Risk Indicators Associated With Permanent Congenital, Delayed-Onset, or Progressive Hearing Loss in Childhood.
-
Caregiver concern [*] regarding hearing, speech, language, or developmental delay ( Roizen, 1999).
-
Family history [*] of permanent childhood hearing loss ( Cone-Wesson et al., 2000; Morton & Nance, 2006).
-
Neonatal intensive care of >5 days, or any of the following regardless of length of stay: ECMO, [*] assisted ventilation, exposure to ototoxic medications (gentamycin and tobramycin) or loop diuretics (furosemide/lasix), and hyperbilirubinemia requiring exchange transfusion ( Fligor et al., 2005; Roizen, 2003).
-
In-utero infections, such as CMV, [*] herpes, rubella, syphilis, and toxoplasmosis ( Fligor et al., 2005; Fowler et al., 1992; Madden et al., 2005; Nance et al., 2006; Pass et al., 2006; Rivera et al., 2002).
-
Craniofacial anomalies, including those involving the pinna, ear canal, ear tags, ear pits, and temporal bone anomalies ( Cone-Wesson et al., 2000).
-
Physical findings, such as white forelock, associated with a syndrome known to include a sensorineural or permanent conductive hearing loss ( Cone-Wesson et al., 2000).
-
Syndromes associated with hearing loss or progressive or late-onset hearing loss, [*] such as neurofibromatosis, osteopetrosis, and Usher syndrome ( Roizen, 2003). Other frequently identified syndromes include Waardenburg, Alport, Pendred, and Jervell and Lange-Nielson ( Nance, 2003).
-
Neurodegenerative disorders, [*] such as Hunter syndrome, or sensory motor neuropathies, such as Friedreich ataxia and Charcot-Marie-Tooth syndrome ( Roizen, 2003).
-
Culture-positive postnatal infections associated with sensorineural hearing loss, [*] including confirmed bacterial and viral (especially herpes viruses and varicella) meningitis ( Arditi et al., 1998; Bess, 1982; Biernath et al., 2006; Roizen, 2003).
-
Head trauma, especially basal skull/temporal bone fracture [*] requiring hospitalization ( Lew et al., 2004; Vartialnen et al., 1985; Zimmerman et al., 1993).
-
Chemotherapy [*] ( Bertolini et al., 2004).
Appendix 2. Algorithm for Hearing Screening. Available at: http://www.medicalhomeinfo.org/screening/Screen%20Materials/Algorithm.pdf
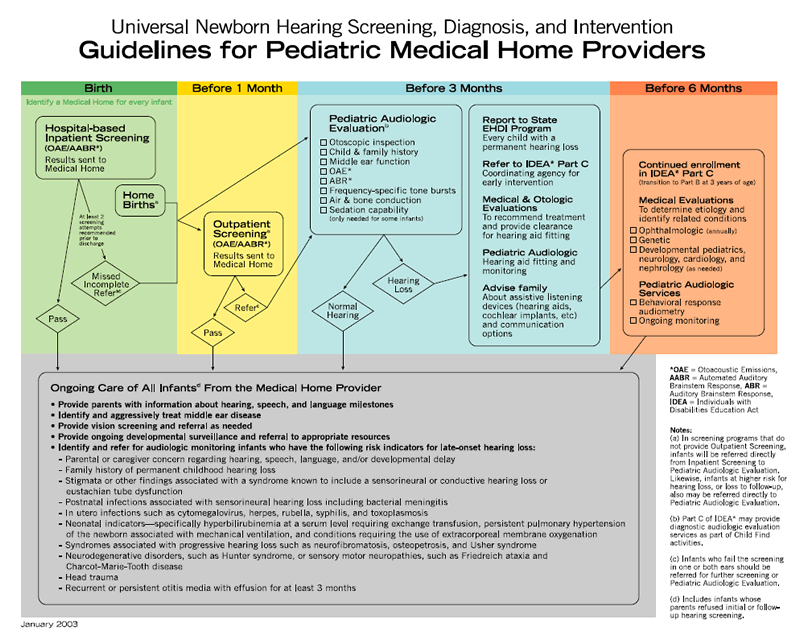
References
Agency for Health Care Policy and Research. (1995). Using clinical practice guidelines to evaluate quality of care. Vol. II: Methods (AHCPR Publication No. 95-0046). Rockville, MD: U.S. Department of Health and Human Services, Public Health Service.
American Academy of Audiology. (2003). Pediatric amplification protocol. Reston, VA: Author. Retrieved January 24, 2007, from www.asha.org/policy/.
American Academy of Pediatrics, Committee on Children With Disabilities. (2005). Care coordination in the medical home: Integrating health and related systems of care for children with special health care needs. Pediatrics, 116, 1238–1244.
American Academy of Pediatrics, Committee on Practice and Ambulatory Medicine. (2000). Recommendations for preventive pediatric health care. Pediatrics, 105, 645–646.
American Academy of Pediatrics, Council on Children With Disabilities, Section on Developmental Behavioral Pediatrics, Bright Futures Steering Committee, Medical Home Initiatives for Children With Special Needs Project Advisory Committee. (2006). Identifying infants and young children with developmental disorders in the medical home: An algorithm for developmental surveillance and screening. Pediatrics, 118, 405–420.
American Academy of Pediatrics, Medical Home Initiatives for Children With Special Needs Project Advisory Committee. (2002). The medical home. Pediatrics, 110, 184–186.
American Academy of Pediatrics, Subcommittee on Otitis Media With Effusion, American Academy of Family Physicians, American Academy of Otolaryngology-Head and Neck Surgery. (2004). Otitis media with effusion. Pediatrics, 113, 1412–1429.
American Academy of Pediatrics, Task Force on Improving the Effectiveness of Newborn Hearing Screening, Diagnosis, and Intervention. (2003). Universal newborn hearing screening, diagnosis, and intervention: Guidelines for pediatric medical home providers. Elk Grove Village, IL: American Academy of Pediatrics. Retrieved January 23, 2007, from http://www.medicalhomeinfo.org/screening/Screen%20Materials/Algorithm.pdf.
American Academy of Pediatrics, Task Force on Newborn and Infant Hearing. (1999). Newborn and infant hearing loss: Detection and intervention. Pediatrics, 103, 527–530.
American Speech-Language-Hearing Association. (1991). The use of FM amplification instruments for infants and preschool children with hearing impairment. Asha 33 (Suppl. 5), 1–2. Retrieved January 24, 2007, from /NR/rdonlyres/226A8C6D-5275-44CC-BFB5-7E0AEA133849/0/18847_1.pdf.
American Speech-Language-Hearing Association. (2004). Guidelines for the audiologic assessment of children from birth to 5 years of age. Rockville, MD: Author. Retrieved January 24, 2007, from www.asha.org/policy/.
American Speech-Language Hearing Association, Joint Committee of ASHA and Council on Education of the Deaf. (1994). Service provision under the Individuals with Disabilities Education Act (IDEA-Part H) to children who are deaf and hard of hearing ages birth to 36 months. Asha, 36, 117–121.
Apuzzo, M. L., & Yoshinaga-Itano, C. (1995). Early identification of infants with significant hearing loss and the Minnesota Child Development Inventory. Seminars in Hearing, 16, 124–137.
Arditi, M., Mason, E. O., Bradley, J. S., Tan, T. O., Barson, W. J., Schutze, G. E., et al. (1998). Three-year multicenter surveillance of pneumococcal meningitis in children: Clinical characteristics, and outcome related to penicillin susceptibility and dexamethasone use. Pediatrics, 102, 1087–1097.
Arehart, K. H., Yoshinaga-Itano, C., Thomson, V., Gabbard, S. A., & Brown, A. S. (1998). State of the states: The status of universal newborn screening, assessment, and intervention systems in 16 states. American Journal of Audiology, 7, 101–114.
Baker-Hawkins, S., & Easterbrooks, S. (1994). Deaf and hard of hearing students: Educational service delivery guidelines. Alexandria, VA: National Association of State Directors of Special Education.
Bamford, J. M. (1998). Early intervention…what then? In F. H. Bess (Ed.), Children with hearing impairment: Contemporary trends (pp. 353–358). Nashville, TN: The Vanderbilt Bill Wilkerson Center Press.
Barbi, M., Binda, S., Caroppo, S., Calvario, A., Germinario, C., Bozzi, A., et al. (2006). Multicity Italian study of congenital cytomegalovirus infection. Pediatric Infectious Diseases Journal, 25, 156–159.
Barrenas, M. L., Jonsson, B., Tuvemo, T., Hellstrom, P. A., & Lundgren, M. (2005). High risk of sensorineural hearing loss in men born small for gestational age with and without obesity or height catch-up growth: A prospective longitudinal register study on birth size in 245,000 Swedish conscripts. Journal of Clinical Endocrinology and Metabolism, 90, 4452–4456.
Benjamini, Y., & Yekutieli, D. (2005). Quantitative trait loci analysis using the false discovery rate. Genetics, 171, 783–790.
Berg, A. L., Spitzer, J. B., Towers, H. M., Bartosiewicz, C., & Diamond, B. E. (2005). Newborn hearing screening in the NICU: Profile of failed auditory brainstem response/passed otoacoustic emission. Pediatrics, 116, 933–938.
Berlin, C. I., Hood, L., Morlet, T., Rose, K., & Brashears, S. (2003). Auditory neuropathy/dys-synchrony: Diagnosis and management. Mental Retardation and Developmental Disabilities Research Reviews, 9, 225–231.
Bertolini, P., Lassalle, M., Mercier, G., Raquin, M. A., Izzi, G., Corradini, N., & Hartmann, O. (2004). Platinum compound-related ototoxicity in children: Long-term follow-up reveals continuous worsening of hearing loss. Journal of Pediatric Hematology/Oncology, 26, 649–655.
Bess, F. H. (1982). Children with unilateral hearing loss. Journal of the Academy of Rehabilitative Audiology, 15, 131–144.
Bess, F. H, Dodd-Murphy, J., & Parker, R. A. (1998). Children with minimal sensorineural hearing loss: Prevalence, educational performance, and functional status. Ear and Hearing, 19, 339–354.
Bess, F. H., & Tharpe, A. M. (1984). Unilateral hearing impairment in children. Pediatrics, 74, 206–216.
Bess, F. H., & Tharpe, A. M. (1986). An introduction to unilateral sensorineural hearing loss in children. Ear and Hearing, 7, 3–13.
Biernath, K. R., Reefhuis, J., Whitney, C. G., Mann, E. A., Costa, P., Eichwald, J., et al. (2006). Bacterial meningitis among children with cochlear implants beyond 24 months after implantation. Pediatrics, 117, 284–289.
Bodner-Johnson, B., & Sass-Lehrer, M. (2003). The young deaf or hard of hearing child. Baltimore: Brookes.
Boppana, S. B., Fowler, K. B., Pass, R. F., Rivera, L. B., Bradford, R. D., Lakeman, F. D., & Britt, W. J. (2005). Congenital cytomegalovirus infection: Association between virus burden in infancy and hearing loss. Journal of Pediatrics, 146, 817–823.
Brick, K. (1999, June). Genetics of deafness, deaf people and the past, present and future . Paper presented at the Workshop on the Genetics of Congenital Hearing Impairment, Atlanta, GA.
Brookhouser, P., Worthington, D., & Kelly, W. (1994). Fluctuating and/or progressive sensorineural hearing loss in children. Laryngoscope, 104, 958–964.
Buchman, C. A., Roush, P. A., Teagle, H. F., Brown, C. J., Zdanski, C. J., & Grose, J. H. (2006). Auditory neuropathy characteristics in children with cochlear nerve deficiency. Ear and Hearing, 27, 399–408.
Calderon, R. (2000). Parental involvement in deaf children's education programs as a predictor of child's language, early reading, and social-emotional development. Journal of Deaf Studies and Deaf Education, 5, 140–155.
Calderon, R., Bargones, J., & Sidman, S. (1998). Characteristics of hearing families and their young deaf and hard of hearing children: Early intervention follow-up. American Annals of the Deaf, 143, 347–362.
Carney, A. E., & Moeller, M. P. (1998). Treatment efficacy: Hearing loss in children. Journal of Speech, Language, and Hearing Research, 41, S61–S84.
Centers for Disease Control and Prevention. (2003). Pneumococcal vaccination for cochlear implant candidates and recipients: Updated recommendations of the Advisory Committee on Immunization Practices. MMWR. Morbidity and Mortality Weekly Report, 52, 739–740.
Clark, T. (1994). SKI*HI: Applications for home-based intervention. In J. Roush & N. D. Matkin (Eds.), Infants and toddlers with hearing loss: Family-centered assessment and intervention (pp. 237–251). Baltimore: York Press.
Cone-Wesson, B., Vohr, B. R., Sininger, Y. S., Widen, J. E., Folsom, R. C., Gorga, M. P., & Norton, S. J. (2000). Identification of neonatal hearing impairment: Infants with hearing loss. Ear and Hearing, 21, 488–507.
D'Agostino, J. A., & Austin, L. (2004). Auditory neuropathy: A potentially under-recognized neonatal intensive care unit sequela. Advances in Neonatal Care, 4, 344–353.
Davis, A., & Hind, S. (2003). The newborn hearing screening programme in England. International Journal of Pediatric Otorhinolaryngology, 67(Suppl. 1), S193–S196.
Denoyelle, F., Marlin, S., Weil, D., Moatti, L., Chauvin, P., Garabedian, E. N., & Petit, C. (1999). Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin-26 gene defect: Implications for genetic counselling. Lancet, 353, 1298–1303.
Diefendorf, A. O., & Gravel, J. S. (1996). Behavioral observation and visual reinforcement audiometry. In S. E. Gerber (Ed.), Handbook of pediatric audiology (pp. 55–83). Washington, DC: Gallaudet University Press.
Doyle, K. J., Burggraaff, B., Fujikawa, S., Kim, J., & MacArthur, C. J. (1997). Neonatal hearing screening with otoscopy, auditory brain stem response, and otoacoustic emissions. Otolaryngology-Head and Neck Surgery, 116, 597–603.
Doyle, K. J., Sininger, Y., & Starr, A. (1998). Auditory neuropathy in childhood. Laryngoscope, 108, 1374–1377.
Eilers, R. E., Miskiel, E., Ozdamar, O., Urbano, R., & Widen, J. E. (1991). Optimization of automated hearing test algorithms: Simulations using an infant response model. Ear and Hearing, 12, 191–198.
Finitzo, T., Albright, K., & O'Neal, J. (1998). The newborn with hearing loss: Detection in the nursery. Pediatrics, 102, 1452–1460.
Fischer, R. M. (1994). The Mama Lere Home: Vanderbilt University. In J. Roush & N. D. Matkin (Eds.), Infants and toddlers with hearing loss: Family-centered assessment and intervention (pp. 195–213). Baltimore: York Press.
Fletcher, R. H., Fletcher, S. W., & Wagner, E. W. (1988). Clinical epidemiology: The essentials (2nd ed.). Baltimore: Williams & Wilkins.
Fligor, B. J., Neault, M. W., Mullen, C. H., Feldman, H. A., & Jones, D. T. (2005). Factors associated with sensorineural hearing loss among survivors of extracorporeal membrane oxygenation therapy. Pediatrics, 115, 1519–1528.
Fortnum, H., & Davis, A. (1997). Epidemiology of permanent childhood hearing impairment in Trent Region, 1985–1993. British Journal of Audiology, 31, 409–446.
Fowler, K., Stagno, S., Pass, R., Britt, W., Boll, T., & Alford, C. (1992). The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. New England Journal of Medicine, 326, 663–667.
Gravel, J., Berg, A., Bradley, M., Cacace, A., Campbell, D., Dalzell, L., et al. (2000). New York State universal newborn hearing screening demonstration project: Effects of screening protocol on inpatient outcome measures. Ear and Hearing, 21, 131–140.
Gravel, J. S., Karma, P., Casselbrant, M. L., Marchisio, P., Andalibi, A., Passali, D, et al. (2005). Recent advances in otitis media. 7. Diagnosis and screening. Annals of Otology, Rhinology & Laryngology Supplement, 194, 104–113.
Herrmann, B. S., Thornton, A. R., & Joseph, J. M. (1995). Automated infant hearing screening using the ABR. Development and validation. American Journal of Audiology, 4, 6–14.
Hochberg, Y., & Benjamini, Y. (1990). More powerful procedures for multiple significance testing. Statistics in Medicine, 9, 811–818.
Holden-Pitt, L., & Diaz, J. (1998). Thirty years of the annual survey of deaf and hard of hearing children and youth: A glance over the decades. American Annals of the Deaf, 143, 72–76.
Hyde, M. D., Sininger, Y. S., & Don, M. (1998). Objective detection and analysis of auditory brainstem response: An historical perspective. Seminars in Hearing, 19, 97–113.
Hyde, M. L., Davidson, M. J., & Alberti, P. W. (1991). Auditory test strategy. In J. T. Jacobson & J. L. Northern (Eds.), Diagnostic audiology (pp. 295–322). Austin, TX: Pro-Ed.
Jacobson, J., & Jacobson, C. (2004). Evaluation of hearing loss in infants and young children. Pediatric Annals, 33, 811–821.
Johnson, D. H. (1999). Deafness and vision disorders: Anatomy and physiology, assessment procedures, ocular anomalies, and educational implications. Springfield, IL: Charles C. Thomas.
Johnson, J. L., White, K. R., Widen, J. E., Gravel, J. S., James, M., Kennalley, T., et al. (2005). A multicenter evaluation of how many infants with permanent hearing loss pass a two-stage otoacoustic emissions/automated auditory brainstem response newborn hearing screening protocol. Pediatrics, 116, 663–672.
Joint Committee on Infant Hearing. (1994a). 1994 position statement. AAO-HNS Bulletin, 12, 13.
Joint Committee on Infant Hearing. (1994b). 1994 position statement. Asha, 36, 38–41.
Joint Committee on Infant Hearing. (2000a). Year 2000 position statement: Principles and guidelines for early hearing detection and intervention programs. American Journal of Audiology, 9, 9–29.
Joint Committee on Infant Hearing. (2000b). Year 2000 position statement: Principles and guidelines for early hearing detection and intervention programs. Pediatrics, 106, 798–817.
Karchmer, M. A., & Allen, T. E. (1999). The functional assessment of deaf and hard of hearing students. American Annals of the Deaf, 144, 68–77.
Keefe, D. H., Gorga, M. P., Neely, S. T., Zhao, F., & Vohr, B. R. (2003). Ear-canal acoustic admittance and reflectance measurements in human neonates. II. Predictions of middle-ear in dysfunction and sensorineural hearing loss. Journal of the Acoustical Society of America, 113, 407–422.
Kennedy, C., McCann, D., Campbell, M. J., Kimm, L., & Thornton, R. (2005). Universal newborn screening for permanent childhood hearing impairment: An 8-year follow-up of a controlled trial. Lancet, 366, 660–662.
Kuhl, P. K., Andruski, J. E., Chistovich, I. A., Chistovich, L. A., Kozhevnikova, E. V., Ryskina, V. L., et al. (1997). Cross-language analysis of phonetic units in language addressed to infants. Science, 277, 684–686.
Kuhl, P. K., Williams, K. A., Lacerda, F., Stevens, K. N., & Lindblom, B. (1992). Linguistic experience alters phonetic perception in infants by 6 months of age. Science, 255, 606–608.
Lew, H. L., Lee, E. H., Miyoshi, Y., Chang, D. G., Date, E. S., & Jerger, J. F. (2004). Brainstem auditory-evoked potentials as an objective tool for evaluating hearing dysfunction in traumatic brain injury. American Journal of Physical Medicine & Rehabilitation, 83, 210–215.
Madden, C., Wiley, S., Schleiss, M., Benton, C., Meinzen-Derr, J., Greinwald, J., & Choo, D. (2005). Audiometric, clinical and educational outcomes in a pediatric symptomatic congenital cytomegalovirus (CMV) population with sensorineural hearing loss. International Journal of Pediatric Otorhinolaryngology, 69, 1191–1198.
Mahshie, S. N. (1995). Educating deaf children bilingually. Washington, DC: Gallaudet University Press.
Marschark, M. (1997). Raising and educating a deaf child. New York: Oxford University Press.
Mason, J. A., & Herrmann, K. R. (1998). Universal infant hearing screening by automated auditory brainstem response measurement. Pediatrics, 101, 221–228.
Mayne, A. M., Yoshinaga-Itano, C., & Sedey, A. L. (1998). Receptive vocabulary development of infants and toddlers who are deaf or hard of hearing. The Volta Review, 100, 29–52.
Mayne, A., Yoshinaga-Itano, C., Sedey, A. L., & Carey, A. (1998). Expressive vocabulary development of infants and toddlers who are deaf or hard of hearing. The Volta Review, 100, 1–28.
McFarland, W. H., Simmons, F. B., & Jones, F. R. (1980). An automated hearing screening technique for newborns. Journal of Speech and Hearing Disorders, 45, 495–503.
Mehl, A. L., & Thomson, V. (1998). Newborn hearing screening: The great omission. Pediatrics, 101(1), E4. Retrieved January 23, 2007, from http://www.pediatrics.org/cgi/content/full/101/1/e4.
Mestan, K. K., Marks, J. D., Hecox, K., Huo, D., & Schreiber, M. D. (2005). Neurodevelopmental outcomes of premature infants treated with inhaled nitric oxide. New England Journal of Medicine, 353, 23–32.
Moeller, M. P. (2000). Early intervention and language development in children who are deaf and hard of hearing. Pediatrics, 106(3), E43. Retrieved January 25, 2007, from http://www.pediatrics.org/cgi/content/full/106/3/e43.
Morton, C. C., & Nance, W. E. (2006). Newborn hearing screening—a silent revolution. New England Journal of Medicine, 354, 2151–2164.
Morzaria, S., Westerberg, B. D., & Kozak, F. K. (2005). Evidence-based algorithm for the evaluation of a child with bilateral sensorineural hearing loss. Journal of Otolaryngology, 34, 297–303.
Nagy, A., Endreffy, E., Streitman, K., Pinter, S., & Pusztai, R. (2004). Incidence and outcome of congenital cytomegalovirus infection in selected groups of preterm and full-term neonates under intensive care. In Vivo, 18, 819–823.
Nance, W. E. (2003). The genetics of deafness. Mental Retardation and Developmental Disabilities Research Reviews, 9, 109–119.
Nance, W. E., & Kearsey, M. J. (2004). Relevance of connexin deafness (DFNB1) to human evolution. American Journal of Human Genetics, 74, 1081–1087.
Nance, W. E., Lim, B. G., & Dodson, K. M. (2006). Importance of congenital cytomegalovirus infections as a cause for pre-lingual hearing loss. Journal of Clinical Virology, 35, 221–225.
National Institute on Deafness and Other Communication Disorders. (1999). Communicating informed consent to individuals who are deaf or hard-of-hearing (NIH Publication No. 00-4689). Bethesda, MD: Author.
National Institutes of Health (1993). Early identification of hearing impairment in infants and young children. NIH Consensus Development Conference Statement. Bethesda, MD: Author. Retrieved January 24, 2007, from http://consensus.nih.gov/1993/1993HearingInfantsChildren092html.htm.
Norton, S. J., Gorga, M. P., Widen, J. E., Folsom, R. C., Sininger, Y., Cone-Wesson, B., et al. (2000). Identification of neonatal hearing impairment: Evaluation of transient evoked otoacoustic emission, distortion product otoacoustic emission, and auditory brain stem response test performance. Ear and Hearing, 21, 508–528.
Orzan, E., Polli, R., Martella, M., Vinanzi, C., Leonardi, M., & Murgia, A. (1999). Molecular genetics applied to clinical practice: The Cx26 hearing impairment. British Journal of Audiology, 33, 291–295.
Ozdamar, O., Delgado, R. E., Eilers, R. E., & Urbano, R. C. (1994). Automated electrophysiologic hearing testing using a threshold-seeking algorithm. Journal of the American Academy of Audiology, 5, 77–88.
Pass, R. F., Fowler, K. B., Boppana, S. B., Britt, W. J., & Stagno, S. (2006). Congenital cytomegalovirus infection following first trimester maternal infection: Symptoms at birth and outcome. Journal of Clinical Virology, 35, 216–220.
Pediatric Working Group. (1996). Amplification for infants and children with hearing loss. American Journal of Audiology, 5, 53–68.
Pipp-Siegel, S., Sedey, A. L., VanLeeuwen, A. M., & Yoshinaga-Itano, C. (2003). Mastery motivation and expressive language in young children with hearing loss. Journal of Deaf Studies and Deaf Education, 8, 133–145.
Pollack, D., Goldberg, D., & Caleffe-Schenck, N. (1997). Educational audiology for the limited-hearing infant and preschooler: An auditory verbal program (3rd ed.). Springfield, IL: Charles C. Thomas.
Pool, K. D., & Finitzo, T. (1989). Evaluation of a computer-automated program for clinical assessment of the auditory brain stem response. Ear and Hearing, 10, 304–310.
Preciado, D. A., Lawson, L., Madden, C., Myer, D., Ngo, C., Bradshaw, J. K., et al. (2005). Improved diagnostic effectiveness with a sequential diagnostic paradigm in idiopathic pediatric sensorineural hearing loss. Otology & Neurotology, 26, 610–615.
Preciado, D. A., Lim, L. H., Cohen, A. P., Madden, C., Myer, D., Ngo, C., et al. (2004). A diagnostic paradigm for childhood idiopathic sensorineural hearing loss. Otolaryngology-Head and Neck Surgery, 131, 804–809.
Prieve, B., Dalzell, L., Berg, A., Bradley, M., Cacace, A., Campbell, D., et al. (2000). The New York State universal newborn hearing screening demonstration project: Outpatient outcome measures. Ear and Hearing, 21, 104–117.
Rance, G. (2005). Auditory neuropathy/dys-synchrony and its perceptual consequences. Trends in Amplification, 9, 1–43.
Rance, G., Cone-Wesson, B., Wunderlich, J., & Dowell, R. (2002). Speech perception and cortical event related potentials in children with auditory neuropathy. Ear and Hearing, 23, 239–253.
Reefhuis, J., Honein, M. A., Whitney, C. G., Chamany, S., Mann, E. A., Biernath, K. R., et al. (2003). Risk of bacterial meningitis in children with cochlear implants. New England Journal of Medicine, 349, 435–445.
Rivera, L. B., Boppana, S. B., Fowler, K. B., Britt, W. J., Stagno, S., & Pass, R. F. (2002). Predictors of hearing loss in children with symptomatic congenital cytomegalovirus infection. Pediatrics, 110, 762–767.
Robertson, C. M., Tyebkhan, J. M., Peliowski, A., Etches, P. C., & Cheung, P. Y. (2006). Ototoxic drugs and sensorineural hearing loss following severe neonatal respiratory failure. Acta Paediatrica, 95, 214–223.
Roizen, N. J. (1999). Etiology of hearing loss in children. Nongenetic causes. Pediatric Clinics of North America, 46, 49–64.
Roizen, N. J. (2003). Nongenetic causes of hearing loss. Mental Retardation and Developmental Disabilities Research Reviews, 9, 120–127.
Rosenfeld, R. M., Culpepper, L., Doyle, K. J., Grundfast, K. M., Hoberman, A., Kenna, M. A., et al. (2004). Clinical practice guideline: Otitis media with effusion. Otolaryngology-Head and Neck Surgery, 130(Suppl. 5), S95–S118.
Roush, J., Bess, F. H., Gravel, J., Harrison, M., Lenihan, S., & Marvelli, A. (2004, February). Preparation of personnel to serve children with hearing loss and their families: Current status and future needs . Paper presented at 2004 Summit on Deafness, Washington, DC.
Sackett, D. L., Hayes, R. B., & Tugwell, P. (1991). Clinical epidemiology: A basic science for clinical medicine (2nd ed.). Boston: Little Brown.
Santos, R. L., Aulchenko, Y. S., Huygen, P. L., van der Donk, K. P., de Wijs, I. J., Kemperman, M. H., et al. (2005). Hearing impairment in Dutch patients with connexin 26 (GJB2) and connexin 30 (GJB6) mutations. International Journal of Pediatric Otorhinolaryngology, 69, 165–174.
Shapiro, S. M. (2003). Bilirubin toxicity in the developing nervous system. Pediatric Neurology, 29, 410–421.
Sharma, A., Tobey, E., Dorman, M., Bharadwaj, S., Martin, K., Gilley, P., & Kunkel, F. (2004). Central auditory maturation and babbling development in infants with cochlear implants. Archives of Otolaryngology-Head & Neck Surgery, 130, 511–516.
Sininger, Y. S., Abdala, C., & Cone-Wesson, B. (1997). Auditory threshold sensitivity of the human neonate as measured by the auditory brainstem response. Hearing Research, 104, 27–38.
Sininger, Y. S., Hood, L. J., Starr, A., Berlin, C. I., & Picton, T. W. (1995). Hearing loss due to auditory neuropathy. Audiology Today, 7, 10–13.
Stapells, D. R., Gravel, J. S., & Martin, B. A. (1995). Thresholds for auditory brain stem responses to tones in notched noise from infants and young children with normal hearing or sensorineural hearing loss. Ear and Hearing, 16, 361–371.
Stark, A. R. American Academy of Pediatrics, Committee on Fetus and Newborn. (2004). Levels of neonatal care. Pediatrics, 114, 1341–1347.
Starr, A., Picton, T. W., Sininger, Y., Hood, L. J., & Berlin, C. I. (1996). Auditory neuropathy. Brain, 119, 741–753.
Starr, A., Sininger, Y. S., & Pratt, H. (2000). The varieties of auditory neuropathy. Journal of Basic and Clinical Physiology and Pharmacology, 11, 215–230.
Thompson, M. (1994). ECHI: The University of Washington, Seattle. In J. Roush & N. D. Natkin (Eds.), Infants and toddlers with hearing loss: Family-centered assessment and intervention (pp. 253–275). Baltimore: York Press.
Traxler, C. B. (2000). The Stanford Achievement Test, 9th Edition: National norming and performance standards for deaf and hard-of-hearing students. Journal of Deaf Studies and Deaf Education, 5, 337–348.
U.S. Department of Health and Human Services, Office of Disease Prevention and Health Promotion. (1991). Healthy People 2000: National health promotion and disease prevention objectives. Washington, DC: U.S. Government Printing Office. Retrieved January 24, 2007, from http://odphp.osophs.dhhs.gov/pubs/hp2000/hppub97.htm.
U.S. Department of Health and Human Services, Office of Disease Prevention and Health Promotion. (2000). Healthy People 2010. Vol. II: Objectives for improving health (2nd ed.). Rockville, MD: Author.
Vartialnen, E., Karjalainen, S., & Karja, J. (1985). Auditory disorders following head injury in children. Acta Oto-Laryngologica, 99, 529–536.
Vohr, B. R., Carty, L. M., Moore, P. E., & Letourneau, K. (1998). The Rhode Island Hearing Assessment Program: Experience with statewide hearing screening (1993–1996). Journal of Pediatrics, 133, 353–357.
Vohr, B. R., Widen, J. E., Cone-Wesson, B., Sininger, Y. S., Gorga, M. P., Folsom, R. C., & Norton, S. J. (2000). Identification of neonatal hearing impairment: Characteristics of infants in the neonatal intensive care unit and well-baby nursery. Ear and Hearing, 21, 373–382.
White, K. (2003). The current status of EHDI programs in the United States. Mental Retardation and Developmental Disabilities Research Reviews, 9, 79–88.
Yoshinaga-Itano, C. (1995). Efficacy of early identification and early intervention. Seminars in Hearing, 16, 115–123.
Yoshinaga-Itano, C. (2001). The social-emotional ramifications of universal newborn hearing screening: Early identification and intervention of children who are deaf or hard of hearing. In Proceedings of the Second International Pediatric Conference: A Sound Foundation Through Early Amplification. Stafa, Switzerland: Phonak. Retrieved January 23, 2007, from http://www.phonak.com/professional/informationpool/proceedings2001.htm.
Yoshinaga-Itano, C. (2003a). Early intervention after universal neonatal hearing screening: Impact on outcomes. Mental Retardation and Developmental Disabilities Research Reviews, 9, 252–266.
Yoshinaga-Itano, C. (2003b). From screening to early identification and intervention: Discovering predictors to successful outcomes for children with significant hearing loss. Journal of Deaf Studies and Deaf Education, 8, 11–30.
Yoshinaga-Itano, C. (2004). Levels of evidence: Universal newborn hearing screening (UNHS) and early hearing detection and intervention systems (EHDI). Journal of Communication Disorders, 37, 451–465.
Yoshinaga-Itano, C., & Abdala de Uzcategui, C. (2001). Early identification and social emotional factors of children with hearing loss and children screened for hearing loss. In E. Kurtzer-White & D. Luterman (Eds.), Early childhood deafness (pp. 13–28). Baltimore: York Press.
Yoshinaga-Itano, C., & Apuzzo, M. L. (1998). The development of deaf and hard of hearing children identified early through the high-risk registry. American Annals of the Deaf, 143, 416–424.
Yoshinaga-Itano, C., & Apuzzo, M. L. (1998). Identification of hearing loss after age 18 months is not early enough. American Annals of the Deaf, 143, 380–387.
Yoshinaga-Itano, C., Coulter, D., & Thomson, V. (2000). The Colorado Newborn Hearing Screening Project: Effects on speech and language development for children with hearing loss. Journal of Perinatology, 20, S132–S137.
Yoshinaga-Itano, C., Coulter, D., & Thomson, V. (2001). Developmental outcomes of children with hearing loss born in Colorado hospitals with and without universal newborn hearing screening programs. Seminars in Neonatology, 6, 521–529.
Yoshinaga-Itano, C., & Sedey, A. (1998). Early speech development in children who are deaf or hard-of-hearing: Interrelationships with language and hearing. The Volta Review, 100, 181–211.
Yoshinaga-Itano, C., Sedey, A. L., Coulter, D. K., & Mehl, A. L. (1998). Language of early- and later-identified children with hearing loss. Pediatrics, 102, 1161–1171.
Zhang, J. H., Chung, T. D., & Oldenburg, K. R. (1999). A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening, 4, 67–73.
Zimmerman, W. D., Ganzel, T. M., Windmill, I. M., Nazar, G. B., & Phillips, M. (1993). Peripheral hearing loss following head trauma in children. Laryngoscope, 103, 87–91.
Notes
[*] Risk indicators that are marked with an asterisk are of greater concern for delayed-onset hearing loss.
Index terms: infants and toddlers, screening, newborns, early intervention
Reference this material as: Joint Committee on Infant Hearing. (2007). Year 2007 position statement: Principles and guidelines for early hearing detection and intervention. Available from www.asha.org/policy.
Reproduced with permission from Pediatrics, 120, 898–921. Copyright © 2007 by the American Academy of Pediatrics.
Disclaimer: The American Speech-Language-Hearing Association disclaims any liability to any party for the accuracy, completeness, or availability of these documents, or for any damages arising out of the use of the documents and any information they contain.
doi:10.1044/policy.PS2007-00281









